| Type: | Package |
| Title: | An R Implementation of the 'Align-GVGD' Method |
| Version: | 0.1.2 |
| Description: | 'Align-GVGD' ('A-GVGD') is a method to predict the impact of 'missense' substitutions based on the properties of amino acid side chains and protein multiple sequence alignments <doi:10.1136/jmg.2005.033878>. 'A-GVGD' is an extension of the original 'Grantham' distance to multiple sequence alignments. This package provides an alternative R implementation to the web version found on http://agvgd.hci.utah.edu/. |
| License: | MIT + file LICENSE |
| Encoding: | UTF-8 |
| RoxygenNote: | 7.2.1 |
| Imports: | crayon, dplyr, glue, grantham, magrittr, purrr, rlang, seqinr, stringr, tibble, tidyr, vctrs |
| Suggests: | rmarkdown, covr, testthat (≥ 3.0.0), |
| Config/testthat/edition: | 3 |
| Depends: | R (≥ 2.10) |
| URL: | https://maialab.org/agvgd/, https://github.com/maialab/agvgd |
| BugReports: | https://github.com/maialab/agvgd/issues |
| NeedsCompilation: | no |
| Packaged: | 2022-09-10 19:30:44 UTC; rmagno |
| Author: | Ramiro Magno  [aut, cre],
Isabel Duarte
[aut],
Ana-Teresa Maia
[aut],
CINTESIS [cph, fnd]
[aut, cre],
Isabel Duarte
[aut],
Ana-Teresa Maia
[aut],
CINTESIS [cph, fnd] |
| Maintainer: | Ramiro Magno <ramiro.magno@gmail.com> |
| Repository: | CRAN |
| Date/Publication: | 2022-09-10 19:42:54 UTC |
agvgd: An R Implementation of the 'Align-GVGD' Method
Description
'Align-GVGD' ('A-GVGD') is a method to predict the impact of 'missense' substitutions based on the properties of amino acid side chains and protein multiple sequence alignments doi:10.1136/jmg.2005.033878. 'A-GVGD' is an extension of the original 'Grantham' distance to multiple sequence alignments. This package provides an alternative R implementation to the web version found on http://agvgd.hci.utah.edu/.
Author(s)
Maintainer: Ramiro Magno ramiro.magno@gmail.com (ORCID)
Authors:
Isabel Duarte iduarte.scientist@gmail.com (ORCID)
Ana-Teresa Maia maia.anateresa@gmail.com (ORCID)
Other contributors:
CINTESIS [copyright holder, funder]
See Also
Useful links:
Report bugs at https://github.com/maialab/agvgd/issues
Pipe operator
Description
See magrittr::%>% for details.
Usage
lhs %>% rhs
Arguments
lhs |
A value or the magrittr placeholder. |
rhs |
A function call using the magrittr semantics. |
Value
The result of calling 'rhs(lhs)'.
Align-GVGD (A-GVGD)
Description
This function implements the Align-GVGD (A-GVGD) method described in Tavtigian et al. (2006).
A-GVGD combines multiple sequence alignment of orthologous sequences with the Grantham distance to classify missense variants, i.e. to distinguish human disease susceptibility missense changes from changes of little clinical significance.
The biochemical variation at each alignment position is converted to a Grantham Variation score (GV) and the difference between these properties and those of the variant amino acid being assessed are calculated and a Grantham Difference score generated (GD). The predicted effect is classed as C0, C15, C25, C35, C45, C55, or C65, with C65 most likely to interfere with function and C0 least likely.
Usage
agvgd(
alignment,
poi,
sub,
mode = c("recycle", "expand_grid"),
sort = FALSE,
keep_self = TRUE,
digits = 2L
)
Arguments
alignment |
A character matrix or an alignment object obtained with
|
poi |
A whole number indicating the position of interest (POI). |
sub |
A character vector of protein residue substitutions to be classified. The amino acids must be provided as one-letter symbols. |
mode |
If both |
sort |
Whether to sort the output by |
keep_self |
Whether to keep those results in the output that correspond
to residues being the same in |
digits |
Integer indicating the number of decimal places to be used in
rounding |
Value
A tibble whose observations refer to the combination alignment position and amino acid substitution; consists of seven variables:
- res
Position of the amino acid residue in the reference protein (first sequence in the alignment). This position corresponds to
poiminus the gaps in the alignment.- poi
Position of interest, i.e. the alignment position at which the amino acid substitution is being assessed.
- ref
Reference amino acid, i.e. the amino acid in the first sequence of the alignment, at the position of interest.
- sub
Amino acid substitution being assessed.
- gv
Grantham variation score.
- gd
Grantham difference score.
- prediction
Predicted effect of the amino acid substitution. This is classed as C0, C15, C25, C35, C45, C55, or C65, with C65 most likely to interfere with function and C0 least likely.
References
Tavtigian, S.V., Deffenbaugh, A. M., Yin, L., Judkins, T., Scholl, T., Samollow, P.B., de Silva, D., Zharkikh, A., Thomas, A. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. Journal of Medical Genetics 43, 295–305 (2006). doi:10.1136/jmg.2005.033878.
Mathe, E., Olivier, M., Kato, S., Ishioka, C., Hainaut, P., Tavtigian, S.V. Computational approaches for predicting the biological effect of p53 missense mutations: a comparison of three sequence analysis based methods. Nucleic Acids Research 34, 1317–1325 (2006). doi:10.1093/nar/gkj518.
Examples
# Read an alignment into R, e.g. the alignment for gene ATM.
alignment_ATM <- read_alignment(gene = 'ATM')
# Predict the impact of changing the first residue (Met) to a Serine (S).
agvgd(alignment = alignment_ATM, poi = 1, sub = 'S')
# `poi` can be a vector of positions, e.g., 3 thru 10, allow for prediction
# of multiple positions at once.
agvgd(alignment = alignment_ATM, poi = 3:10, sub = 'S')
# `poi` expects a position in the frame of reference of the alignment, i.e.
# an alignment position (a column index). However, if you know instead
# the residue position in the reference sequence (first sequence in the
# alignment), then you may use the function `res_to_poi()`
# to convert from residue position to alignment position.
#
# Example: The second residue in the reference sequence of the ATM alignment
# is a Serine, after a Methionine. In the alignment, there is a gap between
# the two residues, so the alignment is 3 but the residue position on the
# protein is 2.
(poi2 <- res_to_poi(alignment_ATM, 2))
agvgd(alignment = alignment_ATM, poi = poi2, sub = 'A')
# Because changes are context-dependent, i.e. they depend on the residue
# variation observed at a given alignment position, the same reference
# residue when replaced with the same substitution will in general have
# a different predicted impact.
agvgd(alignment = alignment_ATM, poi = 9:10, sub = 'S')
# Use the ancillary function `amino_acids()` to get a vector of one-letter
# residue substitutions if you want to quickly assess the impact of all
# possible substitutions.
agvgd(alignment = alignment_ATM, poi = 1, sub = amino_acids())
# Parameter `mode` gives you flexibility on how to combine `poi` and `sub`.
agvgd(alignment = alignment_ATM, poi = 3:4, sub = c('A', 'V'))
# Use 'expand_grid' for all combinations.
agvgd(alignment = alignment_ATM, poi = 3:4, sub = c('A', 'V'), mode = 'expand_grid')
Pre-bundled alignments
Description
This function returns either a data frame of the pre-bundled alignments if
parameter gene is missing (default behaviour), or the file name of the
alignment of a supplied gene name.
Usage
alignment_file(gene)
Arguments
gene |
The gene name of one of the pre-bundled alignments. Run
|
Value
Either a data frame of the pre-bundled alignments if parameter gene
is missing (default behaviour), or the file name of the alignment of a
supplied gene name.
Examples
# List pre-bundled alignment file names and associated genes
alignment_file()
# Retrieve the file name of an alignment
alignment_file("BRCA1")
# You may get the full path to an alignment file with `system.file()`
system.file("extdata", alignment_file("BRCA1"), package = "agvgd")
The 20 standard amino acids
Description
The 20 amino acids that are encoded directly by the codons of the universal genetic code.
Usage
amino_acids(code = c("one_letter", "three_letter"))
Arguments
code |
The type of amino acid symbol to be returned, one-letter ('one_letter') or three-letter ('three_letter') codes. |
Value
A character vector of the 20 standard amino acids.
Examples
# By default `amino_acids` returns one-letter symbols
amino_acids()
# Use code = 'three_letter' instead for three-letter symbols
amino_acids(code = 'three_letter')
Determine CPV ranges
Description
This function determines the range (minimum and maximum) values for the three amino acid side chain property values — composition, polarity and molecular volume — from the amino acids at the alignment position of interest.
The alignment passed in alignment must be an already focused alignment of
three columns whose second column is the position of interest.
Usage
cpv_ranges(alignment, exclude = c("-", "X", NA_character_))
Arguments
alignment |
A character matrix or an alignment object obtained with
|
exclude |
A vector of character values to be ignored when collecting the amino acids at the position of interest. |
Value
A tibble with one single row, of six
variables, i.e., the minimum and maximum values for composition (c_min
and c_max), polarity (p_min and p_max) and molecular volume (v_min
and v_max).
See Also
Examples
# You need to first focus the alignment around the position of interest. The
# position of interest is position 4 in the example below. After subsetting
# the alignment, it becomes position 2.
alignment <- read_alignment('ATM')
alignment[, 3:5]
cpv_ranges(alignment[, 3:5])
# If at the position of interest there are symbols other than amino acid
# symbols, e.g. gaps ("-"), then these are ignored and the calculated ranges
# are based only on the observed amino acids.
alignment[, 270:272]
cpv_ranges(alignment[, 270:272])
Deviation function
Description
This function calculates the deviation in the sense of the Grantham deviation
as introduced by Tavtigian et al. (2006). Essentially, if x lies within the
range [min, max], then dev() returns 0. If x is either below min, or
above max, then dev() returns the absolute difference between x and
min or max, respectively.

Inputs are recycled in the sense of vctrs::vec_recycle().
Usage
dev(x, min, max)
Arguments
x |
A numeric vector. |
min |
A numeric vector. |
max |
A numeric vector. |
Details
Here's a plot showcasing dev() with min = -4 and max = 3:
x <- -10:10; min <- -4; max <- 3 plot(x, y = dev(x, min, max), type = 'l', xlab = 'x', ylab = 'deviation')
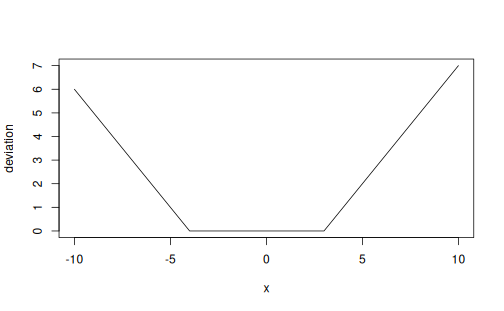
Value
A numeric vector of deviations.
See Also
Examples
# `dev()` returns absolute differences from either min or max (whichever is
# closest).
dev(10, min = -4, max = 4)
dev(-10, min = -4, max = 4)
# `x` can be a vector
dev(-10:10, min = -4, max = 4)
# `min` and `max` can also be vectors, they will be recycled
dev(-10:10, min = -4:16, max = 4:24)
# If `x` contains `NA` values, then `dev()` will return `NA` for
# those cases
dev(c(10, NA), min = -4, max = 4)
# For each calculation of deviation, only either `min` or `max` is used. If
# the unused parameter is `NA` it won't affect the calculation:
dev(c(10, 3), min = c(NA, -4), max = 4)
dev(c(10, -5), min = -4, max = c(4, NA))
Grantham deviation
Description
This function calculates the Grantham deviation (\mathrm{gd}):
\mathrm{gd} = \rho \left((\alpha\ \mathrm{dev}^2(c_x, c_{min}, c_{max}) + \beta\ \mathrm{dev}^2(p_x, p_{min}, p_{max}) + \gamma\ \mathrm{dev}^2(v_x, v_{min}, v_{max})\right)^\frac{1}{2}
where c_x is the value for composition c of amino acid x,
i.e. the atomic weight ratio of hetero (noncarbon) elements in end groups or
rings to carbons in the side chain; p_x is the value for polarity
p of amino acid x; and, v_x is the value for molecular
volume v of amino acid x.
c_x, p_x and v_x are looked up in
grantham::amino_acids_properties based on the amino acid identities passed
in x. The function \mathrm{dev} is implemented in dev(). Remaining
variables in the equation are arguments to gd() and hence are explained
below in the Arguments section.
Usage
gd(
x,
c_min,
c_max,
p_min,
p_max,
v_min,
v_max,
alpha = 1.833,
beta = 0.1018,
gamma = 0.000399,
rho = 50.723
)
Arguments
x |
A character vector of one-letter amino acid codes, indicating missense substitutions. |
c_min |
Amino acid composition, minimum value. |
c_max |
Amino acid, composition, maximum value. |
p_min |
Amino acid polarity, minimum value. |
p_max |
Amino acid polarity, maximum value. |
v_min |
Amino acid molecular volume, maximum value. |
v_max |
Amino acid molecular volume, maximum value. |
alpha |
The constant |
beta |
The constant |
gamma |
The constant |
rho |
Grantham's distances reported in Table 2, Science (1974).
185(4154): 862–4 by R. Grantham, are scaled by a factor (here named
|
Value
A numeric vector of Grantham deviations. Each deviation corresponds
to one of the amino acids indicated in x.
See Also
Examples
gd('S', c_min = 0.39, c_max = 0.74, p_min =4.9, p_max =8.6, v_min = 3, v_max = 32.5)
Grantham variation
Description
This function calculates the Grantham variation (\mathrm{gv}):
\mathrm{gv} = \rho \left((\alpha (c_{max}-c_{min})^2 + \beta (p_{max}-p_{min})^2 + \gamma (v_{max}-v_{min})^2\right)^\frac{1}{2}
The minimum and maximum values are those observed for a set of amino acid residues at the alignment position of interest.
Usage
gv(
c_min,
c_max,
p_min,
p_max,
v_min,
v_max,
alpha = 1.833,
beta = 0.1018,
gamma = 0.000399,
rho = 50.723
)
Arguments
c_min |
Amino acid composition, minimum value. |
c_max |
Amino acid, composition, maximum value. |
p_min |
Amino acid polarity, minimum value. |
p_max |
Amino acid polarity, maximum value. |
v_min |
Amino acid molecular volume, maximum value. |
v_max |
Amino acid molecular volume, maximum value. |
alpha |
The constant |
beta |
The constant |
gamma |
The constant |
rho |
Grantham's distances reported in Table 2, Science (1974).
185(4154): 862–4 by R. Grantham, are scaled by a factor (here named
|
Value
A numeric vector of grantham variation values.
See Also
Examples
# Example based on values from Figure 1C of Tavtigian et al. (2006),
# https://doi.org/10.1136/jmg.2005.033878.
gv(c_min = 0, c_max = 0, p_min = 5.7, p_max = 4.9, v_min = 132, v_max = 105)
Convert an alignment position to residue position
Description
This function converts an alignment position to a position in the frame of the reference protein sequence, i.e., to the positions of the amino acids in the first sequence of the alignment.
Usage
poi_to_res(alignment, poi)
Arguments
alignment |
An alignment. |
poi |
An alignment position. |
Value
An integer vector of positions of the amino acid residues in the reference sequence.
Examples
align_ATM <- read_alignment('ATM')
align_ATM[, 1:5]
# Convert the positions of the first five alignment positions to residue positions
poi_to_res(align_ATM, 1:5)
Converts a sequence profile to an alignment
Description
This function converts a sequence profile as provided in the format of the
package {protean} to an {agvgd} alignment — an alignment in this sense
is simply a character matrix whose elements are protein residues in
one-letter notation, rows are sequences and columns correspond to alignment
positions.
Usage
profile_to_alignment(profile)
Arguments
profile |
A sequence profile object as returned by
|
Value
An alignment object, i.e. a character matrix whose elements are protein residues in one-letter notation. Rows are sequences and columns are alignment positions.
Read in AGVGD Web Results
Description
This function imports into R the results generated by the AGVGD Web application http://agvgd.hci.utah.edu/.
Usage
read_agvgdweb_results(file = stop("`file` is missing"), alignment = NULL)
Arguments
file |
A file path to the results output by the AGVGD Web app. |
alignment |
A character matrix or an alignment object obtained with
|
Value
A tibble of seven columns:
- res
Position of the amino acid residue in the reference protein (first sequence in the alignment). This position corresponds to
poiminus the gaps in the alignment.- poi
Position of interest, i.e. the alignment position at which the amino acid substitution is being assessed. Because this information is not provided by AGVGD Web app this column is always
NA; we keep it though for coherence with the output ofagvgd().- ref
Reference amino acid, i.e. the amino acid in the first sequence of the alignment, at the position of interest.
- sub
Amino acid substitution being assessed.
- gv
Grantham variation score.
- gd
Grantham difference score.
- prediction
Predicted effect of the amino acid substitution. This is classed as C0, C15, C25, C35, C45, C55, or C65, with C65 most likely to interfere with function and C0 least likely.
Read a protein sequence multiple alignment
Description
Reads a protein sequence multiple alignment (PSMA) from either a set of pre-bundled alignments, by gene name, or from a Multi-FASTA file.
Usage
read_alignment(
gene = c("ATM", "BRCA1", "BRCA2", "CHEK2", "MRE11", "MSH6", "NBN", "PALB2", "PMS2",
"RAD50", "RAD51", "XRCC2"),
file = NULL
)
Arguments
gene |
The gene name for which an alignment is provided with this
package. Use the function |
file |
The path to a Multi-FASTA file. If this argument is given, it
takes precedence over the |
Value
An alignment object; essentially, a character matrix, whose elements are protein residues in one-letter notation. Rows are sequences and columns are alignment positions.
Examples
# Read in the alignment for the gene XRCC2
read_alignment('XRCC2')
# Also read in the alignment for the gene XRCC2, but now by specifying
# directly the path to the file.
path <- system.file("extdata", alignment_file("XRCC2"), package = "agvgd")
read_alignment(file = path)
Read a file with amino acid substitutions
Description
This function reads a file with amino acid substitutions. The format of should be the same one as requested by the web version of Align-GVGD.
Usage
read_substitutions(
file = stop("`file` must be specified"),
amino_acid_code = c("one_letter", "three_letter")
)
Arguments
file |
The path to a file with amino acid substitutions. |
amino_acid_code |
The type of symbol used for amino acids in the returned output. |
Value
A tibble listing the amino acids substitutions.
Examples
# "sub.txt" is an example file containing missense substitutions formatted
# according to the requirements indicated in http://agvgd.hci.utah.edu/help.php.
my_file <- system.file("extdata", "sub.txt", package = "agvgd")
cat(readLines(my_file), sep = "\n")
read_substitutions(file = my_file)
# lee2010_sub.txt is a file containing the missense variants studied by
# Lee et al. (2010): https://doi.org/10.1158/0008-5472.CAN-09-4563.
read_substitutions(file = system.file("extdata", "lee2010_sub.txt", package = "agvgd"))
Convert a residue position to an alignment position
Description
This function converts an residue position to a position in the frame of the alignment.
Usage
res_to_poi(alignment, res)
Arguments
alignment |
An alignment. |
res |
A residue position. |
Value
An integer vector of alignment positions corresponding to residue position in the reference sequence.
Examples
align_ATM <- read_alignment('ATM')
align_ATM[, 1:6]
# Convert the positions of the first five residues to alignment positions
res_to_poi(align_ATM, 1:5)
Chop a string into sub-strings of fixed width
Description
Chop a string into sub-strings of fixed width
Usage
str_chop(x, width = 50)
Arguments
x |
A single string. This function is not vectorised, so |
width |
Length of the chopped pieces. |
Value
A character vector of sub-strings of length width.
Export an alignment to FASTA
Description
This function takes an alignment and exports it to a FASTA file.
Usage
write_alignment(alignment, file)
Arguments
alignment |
An alignment. It may be a simple matrix or an object
obtained with |
file |
A file path. |
Value
This function is run for its side effect of writing a file. But it
returns the file path passed in file.
Examples
alignment <- matrix(
c('P', 'M', 'I',
'P', 'I', 'I',
'P', 'L', 'I'),
nrow = 3,
byrow = TRUE
)
# Export an alignment based on a matrix
write_alignment(alignment, "my_alignment.fasta")
cat(readLines("my_alignment.fasta"), sep = "\n")
# Export one of the bundled alignments
write_alignment(read_alignment(gene = 'BRCA1'), "BRCA1.fasta")
cat(readLines("BRCA1.fasta")[1:10], sep = "\n")
Generate and export a list of substitutions
Description
This function exports to a file a list of residue substitutions. The format used will be the same one as requested by the web version of Align-GVGD.
Usage
write_substitutions(
file,
alignment,
poi,
sub,
mode = c("recycle", "expand_grid")
)
Arguments
file |
A file path. |
alignment |
A character matrix or an alignment object obtained with
|
poi |
A whole number indicating the position of interest (POI). |
sub |
A character vector of protein residue substitutions to be classified. The amino acids must be provided as one-letter symbols. |
mode |
If both |
Value
This function is run for its side effect of writing a file. But it
returns the file path passed in file.
Examples
write_substitutions(file = "ex01.csv",
alignment = read_alignment("ATM"),
poi = 20:25,
sub = amino_acids())
cat(readLines("ex01.csv"), sep = "\n")
write_substitutions(file = "ex02.csv",
alignment = read_alignment("ATM"),
poi = 20:21,
sub = amino_acids(),
mode = 'expand_grid')
cat(readLines("ex02.csv"), sep = "\n")