| Type: | Package |
| Title: | Solving Mixed Model Equations in R |
| Version: | 4.4.5 |
| Date: | 2026-03-19 |
| Maintainer: | Giovanny Covarrubias-Pazaran <cova_ruber@live.com.mx> |
| Description: | Structural multivariate-univariate linear mixed model solver for estimation of multiple random effects with unknown variance-covariance structures (e.g., heterogeneous and unstructured) and known covariance among levels of random effects (e.g., pedigree and genomic relationship matrices) (Covarrubias-Pazaran, 2016 <doi:10.1371/journal.pone.0156744>; Maier et al., 2015 <doi:10.1016/j.ajhg.2014.12.006>; Jensen et al., 1997). REML estimates can be obtained using the Direct-Inversion Newton-Raphson and Direct-Inversion Average Information algorithms for the problems r x r (r being the number of records) or using the Henderson-based average information algorithm for the problem c x c (c being the number of coefficients to estimate). Spatial models can also be fitted using the two-dimensional spline functionality available. |
| Depends: | R (≥ 3.5.0), Matrix (≥ 1.1.1), methods, stats, MASS, crayon, enhancer |
| LazyLoad: | yes |
| License: | GPL-2 | GPL-3 [expanded from: GPL (≥ 2)] |
| Imports: | Rcpp (≥ 0.12.19) |
| BugReports: | https://github.com/covaruber/sommer/issues |
| URL: | https://github.com/covaruber/sommer |
| LinkingTo: | Rcpp, RcppArmadillo, RcppProgress |
| Suggests: | rmarkdown, knitr, plyr, parallel, orthopolynom, RSpectra, lattice, testthat (≥ 3.0.0) |
| VignetteBuilder: | knitr |
| Config/testthat/edition: | 3 |
| NeedsCompilation: | yes |
| Packaged: | 2026-03-25 05:24:12 UTC; giovannycovarrubias |
| Author: | Giovanny Covarrubias-Pazaran
 [aut, cre] [aut, cre] |
| Repository: | CRAN |
| Date/Publication: | 2026-03-25 06:10:03 UTC |
Additive relationship matrix
Description
Calculates the realized additive relationship matrix. Currently is the C++ implementation of van Raden (2008).
Usage
A.mat(X,min.MAF=0,return.imputed=FALSE)
Arguments
X |
Matrix ( |
min.MAF |
Minimum minor allele frequency. The A matrix is not sensitive to rare alleles, so by default only monomorphic markers are removed. |
return.imputed |
When TRUE, the imputed marker matrix is returned. |
Details
For vanraden method: the marker matrix is centered by subtracting column means M= X - ms where ms is the coumn means. Then A=M M'/c, where c = \sum_k{d_k}/k, the mean value of the diagonal values of the M M' portion.
Value
If return.imputed = FALSE, the n \times n additive relationship matrix is returned.
If return.imputed = TRUE, the function returns a list containing
- $A
the A matrix
- $X
the imputed marker matrix
References
Endelman, J.B., and J.-L. Jannink. 2012. Shrinkage estimation of the realized relationship matrix. G3:Genes, Genomes, Genetics. 2:1405-1413. doi: 10.1534/g3.112.004259
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Examples
####=========================================####
#### random population of 200 lines with 1000 markers
####=========================================####
X <- matrix(rep(0,200*1000),200,1000)
for (i in 1:200) {
X[i,] <- ifelse(runif(1000)<0.5,-1,1)
}
A <- A.mat(X)
####=========================================####
#### take a look at the Genomic relationship matrix
#### (just a small part)
####=========================================####
# colfunc <- colorRampPalette(c("steelblue4","springgreen","yellow"))
# hv <- heatmap(A[1:15,1:15], col = colfunc(100),Colv = "Rowv")
# str(hv)
Autocorrelation matrix of order 1.
Description
Creates an autocorrelation matrix of order one with parameters specified.
Usage
AR1(x,rho=0.25)
Arguments
x |
vector of the variable to define the factor levels for the AR1 covariance structure. |
rho |
rho value for the matrix. |
Details
Specially useful for constructing covariance structures for rows and ranges to capture better the spatial variation trends in the field. The rho value is assumed fixed and values of the variance component will be optimized through REML.
Value
If everything is defined correctly the function returns:
- $nn
the correlation matrix
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Examples
x <- 1:4
R1 <- AR1(x,rho=.25)
image(R1)
data(DT_sleepstudy, package="enhancer")
DT <- DT_sleepstudy
head(DT)
# define the correlation between Days
D = with(DT, AR1(Days, rho=0.5))
subs = unique(DT$Subject)
# define the correlation between Subjects
S = diag(length(subs))
rownames(S) <- colnames(S) <- subs
# make the kronecker product
DS = kronecker(D,S, make.dimnames = TRUE)
# form the covariance matrix between units
# this is assumes correlation between timepoints
DT$ds <- paste(DT$Days, DT$Subject, sep=":")
DS <- DS[DT$ds, DT$ds]
colnames(DS) <- rownames(DS) <- paste0("u",1:nrow(DS))
# fit the residual model
head(DT)
fm2 <- mmes(Reaction ~ Days,
random= ~ Subject,
rcov = ~vsm(ism(units), Gu=DS), # equivalent to Subject:ar1(Days)
data=DT, tolParInv = 1e-6, verbose = FALSE)
summary(fm2)$varcomp
# the matrix D can take any form: AR1, ARMA, or a custom correlation matrix
Autocorrelation Moving average.
Description
Creates an ARMA matrix of order one with parameters specified.
Usage
ARMA(x, rho=0.25, lambda=0.25)
Arguments
x |
vector of the variable to define the factor levels for the ARMA covariance structure. |
rho |
rho value for the matrix. |
lambda |
dimensions of the square matrix. |
Details
Specially useful for constructing covariance structures for rows and ranges to capture better the spatial variation trends in the field. The rho value is assumed fixed and values of the variance component will be optimized through REML.
Value
If everything is defined correctly the function returns:
- $nn
the correlation matrix
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Examples
x <- 1:4
R1 <- ARMA(x,rho=.25,lambda=0.2)
image(R1)
Compound symmetry matrix
Description
Creates a compound symmetry matrix with parameters specified.
Usage
CS(x, rho=0.25)
Arguments
x |
vector of the variable to define the factor levels for the ARMA covariance structure. |
rho |
rho value for the matrix. |
Details
Specially useful for constructing covariance structures for rows and ranges to capture better the spatial variation trends in the field. The rho value is assumed fixed and values of the variance component will be optimized through REML.
Value
If everything is defined correctly the function returns:
- $nn
the correlation matrix
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Examples
x <- 1:4
R1 <- CS(x,rho=.25)
image(R1)
Dominance relationship matrix
Description
C++ implementation of the dominance matrix. Calculates the realized dominance relationship matrix. Can help to increase the prediction accuracy when 2 conditions are met; 1) The trait has intermediate to high heritability, 2) The population contains a big number of individuals that are half or full sibs (HS & FS).
Usage
D.mat(X,nishio=TRUE,min.MAF=0,return.imputed=FALSE)
Arguments
X |
Matrix ( |
nishio |
If TRUE Nishio ans Satoh. (2014), otherwise Su et al. (2012). See references. |
min.MAF |
Minimum minor allele frequency. The D matrix is not sensitive to rare alleles, so by default only monomorphic markers are removed. |
return.imputed |
When TRUE, the imputed marker matrix is returned. |
Details
The additive marker coefficients will be used to compute dominance coefficients as: Xd = 1-abs(X) for diploids.
For nishio method: the marker matrix is centered by subtracting column means M= Xd - ms where ms is the column means. Then A=M M'/c, where c = 2 \sum_k {p_k (1-p_k)}.
For su method: the marker matrix is normalized by subtracting row means M= Xd - 2pq where 2pq is the product of allele frequencies times 2. Then A=M M'/c, where c = 2 \sum_k {2pq_k (1-2pq_k)}.
Value
If return.imputed = FALSE, the n \times n additive relationship matrix is returned.
If return.imputed = TRUE, the function returns a list containing
- $D
the D matrix
- $imputed
the imputed marker matrix
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Nishio M and Satoh M. 2014. Including Dominance Effects in the Genomic BLUP Method for Genomic Evaluation. Plos One 9(1), doi:10.1371/journal.pone.0085792
Su G, Christensen OF, Ostersen T, Henryon M, Lund MS. 2012. Estimating Additive and Non-Additive Genetic Variances and Predicting Genetic Merits Using Genome-Wide Dense Single Nucleotide Polymorphism Markers. PLoS ONE 7(9): e45293. doi:10.1371/journal.pone.0045293
Examples
####=========================================####
#### EXAMPLE 1
####=========================================####
####random population of 200 lines with 1000 markers
X <- matrix(rep(0,200*1000),200,1000)
for (i in 1:200) {
X[i,] <- sample(c(-1,0,0,1), size=1000, replace=TRUE)
}
D <- D.mat(X)
Epistatic relationship matrix
Description
Calculates the realized epistatic relationship matrix of second order (additive x additive, additive x dominance, or dominance x dominance) using hadamard products with the C++ Armadillo library.
Usage
E.mat(X,nishio=TRUE,type="A#A",min.MAF=0.02)
Arguments
X |
Matrix ( |
nishio |
If TRUE Nishio ans Satoh. (2014), otherwise Su et al. (2012) (see Details in the D.mat help page). |
type |
An argument specifying the type of epistatic relationship matrix desired. The default is the second order epistasis (additive x additive) type="A#A". Other options are additive x dominant (type="A#D"), or dominant by dominant (type="D#D"). |
min.MAF |
Minimum minor allele frequency. The A matrix is not sensitive to rare alleles, so by default only monomorphic markers are removed. |
Details
it is computed as the Hadamard product of the epistatic relationship matrix; E=A#A, E=A#D, E=D#D.
Value
The epistatic relationship matrix is returned.
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Endelman, J.B., and J.-L. Jannink. 2012. Shrinkage estimation of the realized relationship matrix. G3:Genes, Genomes, Genetics. 2:1405-1413. doi: 10.1534/g3.112.004259
Nishio M and Satoh M. 2014. Including Dominance Effects in the Genomic BLUP Method for Genomic Evaluation. Plos One 9(1), doi:10.1371/journal.pone.0085792
Su G, Christensen OF, Ostersen T, Henryon M, Lund MS. 2012. Estimating Additive and Non-Additive Genetic Variances and Predicting Genetic Merits Using Genome-Wide Dense Single Nucleotide Polymorphism Markers. PLoS ONE 7(9): e45293. doi:10.1371/journal.pone.0045293
Examples
####=========================================####
####random population of 200 lines with 1000 markers
####=========================================####
X <- matrix(rep(0,200*1000),200,1000)
for (i in 1:200) {
X[i,] <- sample(c(-1,0,0,1), size=1000, replace=TRUE)
}
E <- E.mat(X, type="A#A")
# if heterozygote markers are present can be used "A#D" or "D#D"
Genome wide association study analysis
Description
THIS FUNCTION IS DEPRECATED. Fits a multivariate/univariate linear mixed model GWAS by likelihood methods (REML), see the Details section below. It uses the mmer function and its core coded in C++ using the Armadillo library to optimize dense matrix operations common in the derect-inversion algorithms. After the model fit extracts the inverse of the phenotypic variance matrix to perform the association test for the "p" markers. Please check the Details section (Model enabled) if you have any issue with making the function run.
The sommer package is updated on CRAN every 3-months due to CRAN policies but you can find the latest source at https://github.com/covaruber/sommer . This can be easily installed typing the following in the R console:
library(devtools)
install_github("covaruber/sommer")
This is recommended since bugs fixes will be immediately available in the GitHub source. For tutorials on how to perform different analysis with sommer please look at the vignettes by typing in the terminal:
vignette("v1.sommer.quick.start")
vignette("v2.sommer.changes.and.faqs")
vignette("v3.sommer.qg")
vignette("v4.sommer.gxe")
or visit https://covaruber.github.io
Usage
GWAS(fixed, random, rcov, data, weights, W,
nIters=20, tolParConvLL = 1e-03, tolParInv = 1e-06,
init=NULL, constraints=NULL,method="NR",
getPEV=TRUE,naMethodX="exclude",
naMethodY="exclude",returnParam=FALSE,
dateWarning=TRUE,date.warning=TRUE,verbose=FALSE,
stepWeight=NULL, emWeight=NULL,
M=NULL, gTerm=NULL, n.PC = 0, min.MAF = 0.05,
P3D = TRUE)
Arguments
fixed |
A formula specifying the response variable(s) and fixed effects, i.e: response ~ covariate for univariate models cbind(response.i,response.j) ~ covariate for multivariate models The fcm function can be used to constrain fixed effects in multi-response models. |
random |
a formula specifying the name of the random effects, i.e. random= ~ genotype + year. Useful functions can be used to fit heterogeneous variances and other special models (see 'Special Functions' in the Details section for more information):
|
rcov |
a formula specifying the name of the error term, i.e. rcov= ~ units. The functions that can be used to fit heterogeneous residual variances are the same used on the random term but the random effect is always "units", i.e. rcov=~vsr(dsr(Location),units) |
data |
a data frame containing the variables specified in the formulas for response, fixed, and random effects. |
weights |
name of the covariate for weights. To be used for the product R = Wsi*R*Wsi, where * is the matrix product, Wsi is the square root of the inverse of W and R is the residual matrix. |
W |
Alternatively, instead of providing a vector of weights the user can specify an entire W matrix (e.g., when covariances exist). To be used first to produce Wis = solve(chol(W)), and then calculate R = Wsi*R*Wsi.t(), where * is the matrix product, and R is the residual matrix. Only one of the arguments weights or W should be used. If both are indicated W will be given the preference. |
nIters |
Maximum number of iterations allowed. Default value is 15. |
tolParConvLL |
Convergence criteria. |
tolParInv |
tolerance parameter for matrix inverse used when singularities are encountered. |
init |
initial values for the variance components. By default this is NULL and variance components are estimated by the method selected, but in case the user want to provide initial values for ALL var-cov components this argument is functional. It has to be provided as a list or an array, where each list element is one variance component and if multitrait model is pursued each element of the list is a matrix of variance covariance components among traits. Initial values can also be provided in the Gt argument of the vsr function.Is highly encouraged to use the Gt and Gtc arguments of the vsr function instead of this argument |
constraints |
when initial values are provided these have to be accompanied by their constraints. See the vsr function for more details on the constraints. Is highly encouraged to use the Gt and Gtc arguments of the vsr function instead of this argument. |
method |
this refers to the method or algorithm to be used for estimating variance components. Direct-inversion Newton-Raphson NR and Average Information AI (Tunnicliffe 1989; Gilmour et al. 1995; Lee et al. 2015). |
getPEV |
a TRUE/FALSE value indicating if the program should return the predicted error variance and variance for random effects. This option is provided since this can take a long time for certain models where p > n by a big extent. |
naMethodX |
one of the two possible values; "include" or "exclude". If "include" is selected then the function will impute the X matrices for fixed effects with the median value. If "exclude" is selected it will get rid of all rows with missing values for the X (fixed) covariates. The default is "exclude". The "include" option should be used carefully. |
naMethodY |
one of the three possible values; "include", "include2" or "exclude". If "include" is selected then the function will impute the response variables with the median value. The difference between "include" and "include2" is only available in the multitrait models when the imputation can happen for the entire matrix of responses or only for complete cases ("include2"). If "exclude" is selected it will get rid of rows in responses where missing values are present for the estimation of variance components. The default is "exclude". |
returnParam |
a TRUE/FALSE value to indicate if the program should return the parameters used for modeling without fitting the model. |
dateWarning |
a TRUE/FALSE value to indicate if the program should warn you when is time to update the sommer package. |
date.warning |
a TRUE/FALSE value to indicate if the program should warn you when is time to update the sommer package. |
verbose |
a TRUE/FALSE value to indicate if the program should return the progress of the iterative algorithm. |
stepWeight |
A vector of values (of length equal to the number of iterations) indicating the weight used to multiply the update (delta) for variance components at each iteration. If NULL the 1st iteration will be multiplied by 0.5, the 2nd by 0.7, and the rest by 0.9. This argument can help to avoid that variance components go outside the parameter space in the initial iterations which doesn't happen very often with the NR method but it can be detected by looking at the behavior of the likelihood. In that case you may want to give a smaller weight to the initial 8-10 iterations. |
emWeight |
A vector of values (of length equal to the number of iterations) indicating with values between 0 and 1 the weight assigned to the EM information matrix. And the values 1 - emWeight will be applied to the NR/AI information matrix to produce a joint information matrix. If NULL weights for EM information matrix are zero and 1 for the NR/AI information matrix. |
M |
The marker matrix containing the marker scores for each level of the random effect selected in the gTerm argument, coded as numeric based on the number of reference alleles in the genotype call, e.g. (-1,0,1) = (aa,Aa,AA), levels in diploid individuals. Individuals in rows and markers in columns. No additional columns should be provided, is a purely numerical matrix. Similar logic applies to polyploid individuals, e.g. (-3,-2,-1,0,1,2,3) = (aaaa,aaaA,aaAA,Aaaa,AAaa,AAAa,AAAA). |
gTerm |
a character vector indicating the random effect linked to the marker matrix M (i.e. the genetic term) in the model. The random effect selected should have the same number of levels than the number of rows of M. When fitting only a random effect without a special covariance structure (e.g., dsr, usr, etc.) you will need to add the call 'u:' to the name of the random effect given the behavior of the naming rules of the solver when having a simple random effect without covariance structure. |
n.PC |
Number of principal components to include as fixed effects. Default is 0 (equals K model). |
min.MAF |
Specifies the minimum minor allele frequency (MAF). If a marker has a MAF less than min.MAF, it is assigned a zero score. |
P3D |
When P3D=TRUE, variance components are estimated by REML only once, without any markers in the model and then a for loop for hypothesis testing is performed. When P3D=FALSE, variance components are estimated by REML for each marker separately. The latter can be quite time consuming. As many models will be run as number of marker. |
Details
Citation
Type citation("sommer") to know how to cite the sommer package in your publications.
Models Enabled
For details about the models enabled and more information about the covariance structures please check the help page of the package (sommer). In general the GWAS model implemented in sommer to obtain marker effect is a generalized linear model of the form:
b = (X'V-X)X'V-y
with X = ZMi
where: b is the marker effect (dimensions 1 x mt) y is the response variable (univariate or multivariate) (dimensions 1 x nt) V- is the inverse of the phenotypic variance matrix (dimensions nt x nt) Z is the incidence matrix for the random effect selected (gTerm argument) to perform the GWAS (dimensions nt x ut) Mi is the ith column of the marker matrix (M argument) (dimensions u x m)
for t traits, n observations, m markers and u levels of the random effect. Depending if P3D is TRUE or FALSE the V- matrix will be calculated once and used for all marker tests (P3D=TRUE) or estimated through REML for each marker (P3D=FALSE).
vignette('sommer.start')
Bug report and contact
If you have any technical questions or suggestions please post it in https://stackoverflow.com or https://stats.stackexchange.com.
If you have any bug report please go to https://github.com/covaruber/sommer or send me an email to address it asap.
Value
If all parameters are correctly indicated the program will return a list with the following information:
Vi |
the inverse of the phenotypic variance matrix V^- = (ZGZ+R)^-1 |
sigma |
a list with the values of the variance-covariance components with one list element for each random effect. |
sigma_scaled |
a list with the values of the scaled variance-covariance components with one list element for each random effect. |
sigmaSE |
Hessian matrix containing the variance-covariance for the variance components. SE's can be obtained taking the square root of the diagonal values of the Hessian. |
Beta |
a data frame for trait BLUEs (fixed effects). |
VarBeta |
a variance-covariance matrix for trait BLUEs |
U |
a list (one element for each random effect) with a data frame for trait BLUPs. |
VarU |
a list (one element for each random effect) with the variance-covariance matrix for trait BLUPs. |
PevU |
a list (one element for each random effect) with the predicted error variance matrix for trait BLUPs. |
fitted |
Fitted values y.hat=XB |
residuals |
Residual values e = Y - XB |
AIC |
Akaike information criterion |
BIC |
Bayesian information criterion |
convergence |
a TRUE/FALSE statement indicating if the model converged. |
monitor |
The values of log-likelihood and variance-covariance components across iterations during the REML estimation. |
scores |
marker scores (-log_(10)p) for the traits |
method |
The method for extimation of variance components specified by the user. |
constraints |
contraints used in the mixed models for the random effects. |
Author(s)
Giovanny Covarrubias-Pazaran
References
Covarrubias-Pazaran G. Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 2016, 11(6): doi:10.1371/journal.pone.0156744
Covarrubias-Pazaran G. 2018. Software update: Moving the R package sommer to multivariate mixed models for genome-assisted prediction. doi: https://doi.org/10.1101/354639
Bernardo Rex. 2010. Breeding for quantitative traits in plants. Second edition. Stemma Press. 390 pp.
Gilmour et al. 1995. Average Information REML: An efficient algorithm for variance parameter estimation in linear mixed models. Biometrics 51(4):1440-1450.
Kang et al. 2008. Efficient control of population structure in model organism association mapping. Genetics 178:1709-1723.
Lee, D.-J., Durban, M., and Eilers, P.H.C. (2013). Efficient two-dimensional smoothing with P-spline ANOVA mixed models and nested bases. Computational Statistics and Data Analysis, 61, 22 - 37.
Lee et al. 2015. MTG2: An efficient algorithm for multivariate linear mixed model analysis based on genomic information. Cold Spring Harbor. doi: http://dx.doi.org/10.1101/027201.
Maier et al. 2015. Joint analysis of psychiatric disorders increases accuracy of risk prediction for schizophrenia, bipolar disorder, and major depressive disorder. Am J Hum Genet; 96(2):283-294.
Rodriguez-Alvarez, Maria Xose, et al. Correcting for spatial heterogeneity in plant breeding experiments with P-splines. Spatial Statistics 23 (2018): 52-71.
Searle. 1993. Applying the EM algorithm to calculating ML and REML estimates of variance components. Paper invited for the 1993 American Statistical Association Meeting, San Francisco.
Yu et al. 2006. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Genetics 38:203-208.
Tunnicliffe W. 1989. On the use of marginal likelihood in time series model estimation. JRSS 51(1):15-27.
Zhang et al. 2010. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 42:355-360.
Examples
####=========================================####
#### For CRAN time limitations most lines in the
#### examples are silenced with one '#' mark,
#### remove them and run the examples using
#### command + shift + C |OR| control + shift + C
####=========================================####
#####========================================####
##### potato example
#####========================================####
#
# data(DT_polyploid, package="enhancer")
# DT <- DT_polyploid
# GT <- GT_polyploid
# MP <- MP_polyploid
# ####=========================================####
# ####### convert markers to numeric format
# ####=========================================####
# numo <- atcg1234(data=GT, ploidy=4);
# numo$M[1:5,1:5];
# numo$ref.allele[,1:5]
#
# ###=========================================####
# ###### plants with both genotypes and phenotypes
# ###=========================================####
# common <- intersect(DT$Name,rownames(numo$M))
#
# ###=========================================####
# ### get the markers and phenotypes for such inds
# ###=========================================####
# marks <- numo$M[common,]; marks[1:5,1:5]
# DT2 <- DT[match(common,DT$Name),];
# DT2 <- as.data.frame(DT2)
# DT2[1:5,]
#
# ###=========================================####
# ###### Additive relationship matrix, specify ploidy
# ###=========================================####
# A <- A.mat(marks)
# ###=========================================####
# ### run it as GWAS model
# ###=========================================####
# ans2 <- GWAS(tuber_shape~1,
# random=~vsr(Name,Gu=A),
# rcov=~units,
# gTerm = "u:Name",
# M=marks, data=DT2)
# plot(ans2$scores[,1])
Combined relationship matrix H
Description
Given a matrix A and a matrix G returns a H matrix with the C++ Armadillo library.
Usage
H.mat(A, G, tau = 1, omega = 1, tolparinv=1e-6)
Arguments
A |
Additive relationship matrix based on pedigree. |
G |
Additive relationship matrix based on marker data. |
tau |
As described by Martini et al. (2018). |
omega |
As described by Martini et al. (2018). |
tolparinv |
Tolerance parameter for matrix inverse used when singularities are encountered in the estimation procedure. |
Details
See references
Value
H Matrix with the relationship between the individuals based on pedigree and corrected by molecular information
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Martini, J. W., Schrauf, M. F., Garcia-Baccino, C. A., Pimentel, E. C., Munilla, S., Rogberg-Munoz, A., ... & Simianer, H. (2018). The effect of the H-1 scaling factors tau and omega on the structure of H in the single-step procedure. Genetics Selection Evolution, 50(1), 16.
Examples
####=========================================####
####random population of 200 lines with 1000 markers
####=========================================####
M <- matrix(rep(0,200*1000),200,1000)
for (i in 1:200) {
M[i,] <- sample(c(-1,0,0,1), size=1000, replace=TRUE)
}
rownames(M) <- 1:nrow(M)
v <- sample(1:nrow(M),100)
M2 <- M[v,]
A <- A.mat(M) # assume this is a pedigree-based matrix for the sake of example
G <- A.mat(M2)
H <- H.mat(A,G)
# colfunc <- colorRampPalette(c("steelblue4","springgreen","yellow"))
# hv <- heatmap(H[1:15,1:15], col = colfunc(100),Colv = "Rowv")
Multivariate Newton-Raphson algorithm
Description
Multivariate Newton-Raphson algorithm used behind mmes when the henderson argument is set to FALSE. Algorithm made available for users that want to avoid the user-friendly interface and provide their matrices directly.
Usage
MNR(Y, # response
X, Gx, # fixed effects
Z, K, # random effects
R, # residual effects
Ge, GeI, # initial values and constraints
W, isInvW, # weights matrix
iters, tolpar, tolparinv, # other params
ai, pev,
verbose, retscaled,
stepweight, emweight
)
Arguments
Y |
A matrix with rows for records and columns for traits. Expected class is a 'matrix'. |
X |
Design matrices for fixed effects. Expected class is a 'list' with as many design matrices as fixed effects. Matrices within the list should be of class 'matrix' |
Gx |
Trait multiplier matrix for fixed effects. Expected class is a 'list' with as many matrices as fixed effects. Each matrix is a square matrix with as many rows and columns as number of traits. Each matrix is of class 'matrix' |
Z |
Design matrices for random effects. Expected class is a 'list' with as many design matrices as random effects. Each element in the list should be a matrix of class 'dgCMatrix' |
K |
Covariance matrices for design matrices of random effects. Expected class is a 'list' with as many covariance matrices as random effects specified in Z. Each element in the list should be a matrix of class 'dgCMatrix' |
R |
Residual matrices for residual effects. Expected class is a 'list' with as many residual matrices as residual effects. Each element in the list should be a square matrix of dimensions n x n, where n is the number of records. Each matrix should be of class 'matrix' |
Ge |
Initial values for variance components. Expected class is a 'list' with as many variance component matrices as random effects specified in Z. Each element in the list should be a matrix of dimensions t x t, where t is the number of traits and of class 'matrix'. Initial values are any real values. |
GeI |
Initial constraints for variance components. Expected class is a 'list' with as many variance component constraints matrices as random effects specified in Z. Each element in the list should be a matrix of dimensions t x t, where t is the number of traits and of class 'matrix'. Values expected are: 0: not to be estimated 1: estimated and constrained to be positive (i.e. variance component) 2: estimated and unconstrained (can be negative or positive, i.e. covariance component) 3: not to be estimated but fixed (value has to be provided in the Gti argument) Please notice that lower triangular values of these matrices have to be equal to zero since only the values in the upper triangular are estimated. |
W |
Design matrix for weights. A matrix of class 'matrix' for weighting the records. |
isInvW |
A value of class 'logical' to indicate if the W matrix provided is already an inverse or not. This aims to speed up the computations. |
iters |
Maximum number of iterations allowed in REML. Value is of class 'integer'. |
tolpar |
Convergence criteria for the change in log-likelihood. Value is of class 'numeric'. |
tolparinv |
Tolerance parameter for matrix inverse used when singularities are encountered in the estimation procedure. Value is of class 'numeric'. |
ai |
A value of class 'logical' to indicate if the Average Information algorithm should be used instead. That is faster but much less stable. |
pev |
A value of class 'logical' to indicate if the predicted error variance should be computed or not. If FALSE computations are speeded up for models with many effects to be estimated. |
verbose |
A value of class 'logical' to value to indicate if the program should return the progress of the iterative algorithm. |
retscaled |
A value of class 'logical' to indicate if we should avoid scaling the traits. |
stepweight |
Vector of class 'numeric' with length equal to niters to specify the relative weight given to the second derivative Newton update. |
emweight |
Vector of class 'numeric' with length equal to niters to specify the relative weight given to the EM update compared to the NR update. |
Details
This is the Rcpp-coded Direct-Inversion LMM REML algorithm used behind the mmer function.
Value
If all parameters are correctly indicated the program will return a list with the following information:
Vi |
the inverse of the phenotypic variance matrix V^- = (ZGZ+R)^-1 |
P |
the projection matrix Vi - [Vi*(X*Vi*X)^-*Vi] |
sigma |
a list with the values of the variance-covariance components with one list element for each random effect. |
sigma_scaled |
a list with the values of the scaled variance-covariance components with one list element for each random effect. |
sigmaSE |
Hessian matrix containing the variance-covariance for the variance components. SE's can be obtained taking the square root of the diagonal values of the Hessian. |
Beta |
a data frame for trait BLUEs (fixed effects). |
VarBeta |
a variance-covariance matrix for trait BLUEs |
U |
a list (one element for each random effect) with a data frame for trait BLUPs. |
VarU |
a list (one element for each random effect) with the variance-covariance matrix for trait BLUPs. |
PevU |
a list (one element for each random effect) with the predicted error variance matrix for trait BLUPs. |
fitted |
Fitted values y.hat=XB |
residuals |
Residual values e = Y - XB |
AIC |
Akaike information criterion |
BIC |
Bayesian information criterion |
convergence |
a TRUE/FALSE statement indicating if the model converged. |
monitor |
The values of log-likelihood and variance-covariance components across iterations during the REML estimation. |
percChange |
The percent change of variance components across iterations. There should be one column less than the number of iterations. Calculated as percChange = ((x_i/x_i-1) - 1) * 100 where i is the ith iteration. |
dL |
The vector of first derivatives of the likelihood with respect to the ith variance-covariance component. |
dL2 |
The matrix of second derivatives of the likelihood with respect to the i.j th variance-covariance component. |
References
Mrode, R. A. (2014). Linear models for the prediction of animal breeding values. Cabi.
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
See Also
mmes – the core function of the package
Examples
data(DT_cpdata, package="enhancer")
DT <- DT_cpdata
# response matrix
y <- cbind(imputev(DT$Yield), imputev(DT$Firmness))
# fixed effect incidence matrix
X <- model.matrix(~Rowf, data=DT)
# random effect incidence matrix
Z <- Matrix::sparse.model.matrix(~id - 1, data=DT)
colnames(Z) <- gsub("id","",colnames(Z))
# covariance for random effect
GT <- GT_cpdata
A <- A.mat(GT) # additive relationship matrix
A <- A[colnames(Z),colnames(Z)] # make sure of the order
A <- A + diag(1e-4,nrow(A), nrow(A))
# residual effect incidence matrix (dimensions equal nrow(y))
R1 <- Matrix::Diagonal(n=nrow(y))
# weights matrix (dimensions equal nrow(y))
W <- diag(nrow(y))
# some other parameters
maxIter=3
stepWeight <- rep(0.9, maxIter)
stepWeight[1:2] <- c(0.5, 0.7)
emWeights <- rep(0,maxIter)
# model fit
res <- MNR( Y=y, # multi-trait response
X=list(X), Gx=list(diag(2)), # fixed effects
Z=list(Z), K=list(A), # random effects
R=list(R1), # residual effects
Ge=list( (diag(2)*.3)+.15 , diag(2)*0.75 ), # inital vc
GeI=list(unsm2(2),diag(2)), # vc constraints
W=W, isInvW=TRUE, # weights for records
iters=maxIter,
tolpar=1e-4, tolparinv=1e-6, # tolerance
ai=FALSE, pev=FALSE, # algorithm specifics
verbose=TRUE,
retscaled=FALSE,
stepweight=stepWeight, # second derivatives weights
emweight=emWeights # em update weights
)
res$sigma
res$Beta
res$U[[1]]
anova form a GLMM fitted with mmes
Description
anova method for class "mmes".
Usage
## S3 method for class 'mmes'
anova(object, object2=NULL, ...)
Arguments
object |
an object of class |
object2 |
an object of class |
... |
Further arguments to be passed |
Value
vector of anova
Author(s)
Giovanny Covarrubias
See Also
atm covariance structure
Description
atm creates a diagonal covariance structure for specific levels of the random effect to be used with the mmes solver.
Usage
atm(x, levs, thetaC, theta)
Arguments
x |
vector of observations for the random effect. |
levs |
levels of the random effect to use for building the incidence matrices. |
thetaC |
an optional symmetric matrix for constraints in the variance-covariance components. The symmetric matrix should have as many rows and columns as the number of levels in the factor 'x'. The values in the matrix define how the variance-covariance components should be estimated: 0: component will not be estimated 1: component will be estimated and constrained to be positive 2: component will be estimated and unconstrained 3: component will be fixed to the value provided in the theta argument |
theta |
an optional symmetric matrix for initial values of the variance-covariance components. The symmetric matrix should have as many rows and columns as the number of levels in the factor 'x'. The values in the matrix define the initial values of the variance-covariance components that will be subject to the constraints provided in thetaC. If not provided, initial values will be calculated as: theta* = diag(ncol(mm))*.05 + matrix(.1,ncol(mm),ncol(mm)) where mm is the incidence matrix for the factor 'x'. The values provided should be scaled by the variance of the response variable. theta = theta*/var(y) |
Value
- $res
a list with the provided vector and the variance covariance structure expected.
Author(s)
Giovanny Covarrubias-Pazaran
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Examples
x <- as.factor(c(1:5,1:5,1:5));x
atm(x, c("1","2"))
## how to use the theta and thetaC arguments:
# data(DT_example, package="enhancer")
# DT <- DT_example
# theta <- diag(2)*2; theta # initial VCs
# thetaC <- diag(2)*3; thetaC # fixed VCs
# ans1 <- mmes(Yield~Env,
# random= ~ vsm( atm(Env, levs=c("CA.2013", "CA.2011"),
# theta = theta,thetaC = thetaC),ism(Name) ),
# rcov= ~ units, nIters = 1,
# data=DT)
# summary(ans1)$varcomp
coef form a GLMM fitted with mmes
Description
coef method for class "mmes".
Usage
## S3 method for class 'mmes'
coef(object, ...)
Arguments
object |
an object of class |
... |
Further arguments to be passed |
Value
vector of coef
Author(s)
Giovanny Covarrubias
See Also
Imputing a matrix using correlations
Description
corImputation imputes missing data based on the correlation that exists between row levels.
Usage
corImputation(wide, Gu=NULL, nearest=10, roundR=FALSE)
Arguments
wide |
numeric matrix with individuals in rows and time variable in columns (e.g., environments, genetic markers, etc.). |
Gu |
optional correlation matrix between the individuals or row levels. If NULL it will be computed as the correlation of t(wide). |
nearest |
integer value describing how many nearest neighbours (the ones showing the highest correlation) should be used to average and return the imputed value. |
roundR |
a TRUE/FALSE statement describing if the average result should be rounded or not. This may be specifically useful for categorical data in the form of numbers (e.g., -1,0,1). |
Value
- $res
a list with the imputed matrix and the original matrix.
Author(s)
Giovanny Covarrubias-Pazaran
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Examples
####################################
### imputing genotype data example
####################################
# data(DT_cpdata, package="enhancer")
# X <- GT_cpdata
# # add missing data
# v <- sample(1:length(X), 500)
# Xna <- X
# Xna[v]<- NA
# ## impute (can take some time)
# Y <- corImputation(wide=Xna, Gu=NULL, nearest=20, roundR=TRUE)
# cm <- table(Y$imputed[v],X[v])
# ## calculate accuracy
# sum(diag(cm))/length(v)
####################################
### imputing phenotypic data example
####################################
# data(DT_h2, package="enhancer")
# X <- reshape(DT_h2[,c("Name","Env","y")], direction = "wide", idvar = "Name",
# timevar = "Env", v.names = "y", sep= "_")
# rownames(X) <- X$Name
# X <- as.matrix(X[,-1])
# head(X)
# # add missing data
# v <- sample(1:length(X), 50)
# Xna <- X
# Xna[v]<- NA
# ## impute
# Y <- corImputation(wide=Xna, Gu=NULL, nearest=20, roundR=TRUE)
# plot(y=Y$imputed[v],x=X[v], xlab="true",ylab="predicted")
# cor(Y$imputed[v],X[v], use = "complete.obs")
covariance between random effects
Description
covm merges the incidence matrices and covariance matrices of two random effects to fit an unstructured model between 2 different random effects to be fitted with the mmes solver.
Usage
covm(ran1, ran2, thetaC=NULL, theta=NULL)
Arguments
ran1 |
the random call of the first random effect. |
ran2 |
the random call of the first random effect. |
thetaC |
an optional matrix for constraints in the variance components. |
theta |
an optional symmetric matrix for initial values of the variance-covariance components. When providing customized values, these values should be scaled with respect to the original variance. For example, to provide an initial value of 1 to a given variance component, theta would be built as: theta = matrix( 1 / var(response) ) The symmetric matrix should have as many rows and columns as the number of levels in the factor 'x'. The values in the matrix define the initial values of the variance-covariance components that will be subject to the constraints provided in thetaC. If not provided, initial values will be calculated as: theta = diag(ncol(mm))*.05 + matrix(.1,ncol(mm),ncol(mm)) where mm is the incidence matrix for the factor 'x'. |
Details
This implementation aims to fit models where covariance between random variables is expected to exist. For example, indirect genetic effects.
Value
- $Z
a incidence matrix Z* = Z Gamma which is the original incidence matrix for the timevar multiplied by the loadings.
Author(s)
Giovanny Covarrubias-Pazaran
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Bijma, P. (2014). The quantitative genetics of indirect genetic effects: a selective review of modelling issues. Heredity, 112(1), 61-69.
See Also
The function vsm to know how to use covm in the mmes solver.
Examples
data(DT_ige, package="enhancer")
DT <- DT_ige
covRes <- with(DT, covm( vsm(ism(focal)) , vsm(ism(neighbour)) ) )
str(covRes)
# look at DT_ige help page to see how to fit an actual model
customized covariance structure
Description
csm creates a customized covariance structure for specific levels of the random effect to be used with the mmes solver.
Usage
csm(x, mm, thetaC, theta)
Arguments
x |
vector of observations for the random effect. |
mm |
customized variance-covariance structure for the levels of the random effect. |
thetaC |
an optional symmetric matrix for constraints in the variance-covariance components. The symmetric matrix should have as many rows and columns as the number of levels in the factor 'x'. The values in the matrix define how the variance-covariance components should be estimated: 0: component will not be estimated 1: component will be estimated and constrained to be positive 2: component will be estimated and unconstrained 3: component will be fixed to the value provided in the theta argument |
theta |
an optional symmetric matrix for initial values of the variance-covariance components. When providing customized values, these values should be scaled with respect to the original variance. For example, to provide an initial value of 1 to a given variance component, theta would be built as: theta = matrix( 1 / var(response) ) The symmetric matrix should have as many rows and columns as the number of levels in the factor 'x'. The values in the matrix define the initial values of the variance-covariance components that will be subject to the constraints provided in thetaC. If not provided, initial values will be calculated as: theta = diag(ncol(mm))*.05 + matrix(.1,ncol(mm),ncol(mm)) where mm is the incidence matrix for the factor 'x'. |
Value
- $res
a list with the provided vector and the variance covariance structure expected for the levels of the random effect.
Author(s)
Giovanny Covarrubias-Pazaran
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
See Also
The function vsm to know how to use csm in the mmes solver.
Examples
x <- as.factor(c(1:5,1:5,1:5));x
csm(x,matrix(1,5,5))
data frame to matrix
Description
This function takes a matrix that is in data frame format and transforms it into a matrix. Other packages that allows you to obtain an additive relationship matrix from a pedigree is the 'pedigreemm' package.
Usage
dfToMatrix(x, row="Row",column="Column",
value="Ainverse", returnInverse=FALSE,
bend=1e-6)
Arguments
x |
ginv element, output from the Ainverse function. |
row |
name of the column in x that indicates the row in the original relationship matrix. |
column |
name of the column in x that indicates the column in the original relationship matrix. |
value |
name of the column in x that indicates the value for a given row and column in the original relationship matrix. |
returnInverse |
a TRUE/FALSE value indicating if the inverse of the x matrix should be computed once the data frame x is converted into a matrix. |
bend |
a numeric value to add to the diagonal matrix in case matrix is singular for inversion. |
Value
K |
pedigree transformed in a relationship matrix. |
Kinv |
inverse of the pedigree transformed in a relationship matrix. |
Author(s)
Giovanny Covarrubias-Pazaran
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Examples
library(Matrix)
m <- matrix(1:9,3,3)
m <- tcrossprod(m)
mdf <- as.data.frame(as.table(m))
mdf
dfToMatrix(mdf, row = "Var1", column = "Var2",
value = "Freq",returnInverse=FALSE )
diagonal covariance structure
Description
dsm creates a diagonal covariance structure for the levels of the random effect to be used with the mmes solver.
Usage
dsm(x, thetaC=NULL, theta=NULL)
Arguments
x |
vector of observations for the random effect. |
thetaC |
an optional symmetric matrix for constraints in the variance-covariance components. The symmetric matrix should have as many rows and columns as the number of levels in the factor 'x'. The values in the matrix define how the variance-covariance components should be estimated: 0: component will not be estimated 1: component will be estimated and constrained to be positive 2: component will be estimated and unconstrained 3: component will be fixed to the value provided in the theta argument |
theta |
an optional symmetric matrix for initial values of the variance-covariance components. When providing customized values, these values should be scaled with respect to the original variance. For example, to provide an initial value of 1 to a given variance component, theta would be built as: theta = matrix( 1 / var(response) ) The symmetric matrix should have as many rows and columns as the number of levels in the factor 'x'. The values in the matrix define the initial values of the variance-covariance components that will be subject to the constraints provided in thetaC. If not provided, initial values will be calculated as: diag(ncol(mm))*.05 + matrix(.1,ncol(mm),ncol(mm)) where mm is the incidence matrix for the factor 'x'. |
Value
- $res
a list with the provided vector and the variance covariance structure expected for the levels of the random effect.
Author(s)
Giovanny Covarrubias-Pazaran
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
See Also
See the function vsm to know how to use dsm in the mmes solver.
Examples
x <- as.factor(c(1:5,1:5,1:5));x
dsm(x)
## how to use the theta and thetaC arguments:
# data(DT_example, package="enhancer")
# DT <- DT_example
# theta <- diag(3)*2; theta # initial VCs
# thetaC <- diag(3)*3; thetaC # fixed VCs
# ans1 <- mmes(Yield~Env,
# random= ~ vsm( dsm(Env,theta = theta,thetaC = thetaC),ism(Name) ),
# rcov= ~ units,
# data=DT)
# summary(ans1)$varcomp
fitted form a LMM fitted with mmes
Description
fitted method for class "mmes".
Usage
## S3 method for class 'mmes'
fitted(object, ...)
Arguments
object |
an object of class |
... |
Further arguments to be passed to the mmes function |
Value
vector of fitted values of the form y.hat = Xb + Zu including all terms of the model.
Author(s)
Giovanny Covarrubias
See Also
Examples
# data(DT_cpdata, package="enhancer")
# DT <- DT_cpdata
# GT <- GT_cpdata
# MP <- MP_cpdata
# #### create the variance-covariance matrix
# A <- A.mat(GT) # additive relationship matrix
# #### look at the data and fit the model
# head(DT)
# mix1 <- mmes(Yield~1,
# random=~vsm(ism(id),Gu=A)
# + Rowf + Colf + spl2Dc(Row,Col),
# rcov=~units,
# data=DT)
#
# ff=fitted(mix1)
#
# colfunc <- colorRampPalette(c("steelblue4","springgreen","yellow"))
# lattice::wireframe(`u:Row.fitted`~Row*Col, data=ff$dataWithFitted,
# aspect=c(61/87,0.4), drape=TRUE,# col.regions = colfunc,
# light.source=c(10,0,10))
# lattice::levelplot(`u:Row.fitted`~Row*Col, data=ff$dataWithFitted, col.regions = colfunc)
fixed indication matrix
Description
fixm creates a square matrix with 3's in the diagnals and off-diagonals to quickly specify a fixed constraint in the Gtc argument of the vsm function.
Usage
fixm(x, reps=NULL)
Arguments
x |
integer specifying the number of traits to be fitted for a given random effect. |
reps |
integer specifying the number of times the matrix should be repeated in a list format to provide easily the constraints in complex models that use the ds(), us() or cs() structures. |
Value
- $res
a matrix or a list of matrices with the constraints to be provided in the Gtc argument of the
vsmfunction.
Author(s)
Giovanny Covarrubias-Pazaran
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Examples
fixm(4)
fixm(4,2)
identity covariance structure
Description
ism creates an identity covariance structure for the levels of the random effect to be used with the mmes solver. Any random effect with a special covariance structure should end with an ism() structure.
Usage
ism(x, thetaC=NULL, theta=NULL)
Arguments
x |
vector of observations for the random effect. |
thetaC |
an optional 1 x 1 matrix for constraints in the variance-covariance components. The values in the matrix define how the variance-covariance components should be estimated: 0: component will not be estimated 1: component will be estimated and constrained to be positive (default) 2: component will be estimated and unconstrained 3: component will be fixed to the value provided in the theta argument |
theta |
an optional 1 x 1 matrix for initial values of the variance-covariance component. When providing customized values, these values should be scaled with respect to the original variance. For example, to provide an initial value of 1 to a given variance component, theta would be built as: theta = matrix( 1 / var(response) ) The values in the matrix define the initial values of the variance-covariance components that will be subject to the constraints provided in thetaC. If not provided, initial values (theta) will be 0.15 |
Value
- $res
a list with the provided vector and the variance covariance structure expected for the levels of the random effect.
Author(s)
Giovanny Covarrubias-Pazaran
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
See Also
See the function vsm to know how to use ism in the mmes solver.
Examples
x <- as.factor(c(1:5,1:5,1:5));x
ism(x)
# data(DT_example, package="enhancer")
# ans1 <- mmes(Yield~Env,
# random= ~ vsm( ism( Name ) ),
# data=DT_example)
# summary(ans1)$varcomp
mixed model equations for r records
Description
The mmer function uses the Direct-Inversion Newton-Raphson or Average Information coded in C++ using the Armadillo library to optimize dense matrix operations common in genomic selection models. These algorithms are intended to be used for problems of the type c > r (more coefficients to estimate than records in the dataset) and/or dense matrices. For problems with sparse data, or problems of the type r > c (more records in the dataset than coefficients to estimate), the MME-based algorithm in the mmes function is faster and we recommend to shift to use that function.
Usage
mmer(fixed, random, rcov, data, weights, W, nIters=20, tolParConvLL = 1e-03,
tolParInv = 1e-06, init=NULL, constraints=NULL,method="NR", getPEV=TRUE,
naMethodX="exclude", naMethodY="exclude",returnParam=FALSE,
dateWarning=TRUE,date.warning=TRUE,verbose=TRUE, reshapeOutput=TRUE, stepWeight=NULL,
emWeight=NULL, contrasts=NULL)
Arguments
fixed |
A formula specifying the response variable(s) and fixed effects, e.g.: response ~ covariate for univariate models cbind(response.i,response.j) ~ covariate for multivariate models The |
random |
A formula specifying the name of the random effects, e.g. random= ~ genotype + year. Useful functions can be used to fit heterogeneous variances and other special models (see 'Special Functions' in the Details section for more information):
** ** **
|
rcov |
A formula specifying the name of the error term, e.g., rcov= ~ units. Special heterogeneous and special variance models and constraints for the residual part are the same used on the random term but the name of the random effect is always "units" which can be thought as a column with as many levels as rows in the data, e.g., rcov=~vsr(dsr(covariate),units) |
data |
A data frame containing the variables specified in the formulas for response, fixed, and random effects. |
weights |
Name of the covariate for weights. To be used for the product R = Wsi*R*Wsi, where * is the matrix product, Wsi is the square root of the inverse of W and R is the residual matrix. |
W |
Alternatively, instead of providing a vector of weights the user can specify an entire W matrix (e.g., when covariances exist). To be used first to produce Wis = solve(chol(W)), and then calculate R = Wsi*R*Wsi.t(), where * is the matrix product, and R is the residual matrix. Only one of the arguments weights or W should be used. If both are indicated W will be given the preference. |
nIters |
Maximum number of iterations allowed. |
tolParConvLL |
Convergence criteria for the change in log-likelihood. |
tolParInv |
Tolerance parameter for matrix inverse used when singularities are encountered in the estimation procedure. |
init |
Initial values for the variance components. By default this is NULL and initial values for the variance components are provided by the algorithm, but in case the user want to provide initial values for ALL var-cov components this argument is functional. It has to be provided as a list, where each list element corresponds to one random effect (1x1 matrix) and if multitrait model is pursued each element of the list is a matrix of variance covariance components among traits for such random effect. Initial values can also be provided in the Gti argument of the vsr function. Is highly encouraged to use the Gti and Gtc arguments of the vsr function instead of this argument, but these argument can be used to provide all initial values at once |
constraints |
When initial values are provided these have to be accompanied by their constraints. See the vsr function for more details on the constraints. Is highly encouraged to use the Gti and Gtc arguments of the vsr function instead of this argument but these argument can be used to provide all constraints at once. |
method |
This refers to the method or algorithm to be used for estimating variance components. Direct-inversion Newton-Raphson NR and Average Information AI (Tunnicliffe 1989; Gilmour et al. 1995; Lee et al. 2015). |
getPEV |
A TRUE/FALSE value indicating if the program should return the predicted error variance and variance for random effects. This option is provided since this can take a long time for certain models where p is > n by a big extent. |
naMethodX |
One of the two possible values; "include" or "exclude". If "include" is selected then the function will impute the X matrices for fixed effects with the median value. If "exclude" is selected it will get rid of all rows with missing values for the X (fixed) covariates. The default is "exclude". The "include" option should be used carefully. |
naMethodY |
One of the three possible values; "include", "include2" or "exclude" (default) to treat the observations in response variable to be used in the estimation of variance components. The first option "include" will impute the response variables for all rows with the median value, whereas "include2" imputes the responses only for rows where there is observation(s) for at least one of the responses (only available in the multi-response models). If "exclude" is selected (default) it will get rid of rows in response(s) where missing values are present for at least one of the responses. |
returnParam |
A TRUE/FALSE value to indicate if the program should return the parameters to be used for fitting the model instead of fitting the model. |
dateWarning |
A TRUE/FALSE value to indicate if the program should warn you when is time to update the sommer package. |
date.warning |
A TRUE/FALSE value to indicate if the program should warn you when is time to update the sommer package. This argument will be removed soon, just left for backcompatibility. |
verbose |
A TRUE/FALSE value to indicate if the program should return the progress of the iterative algorithm. |
reshapeOutput |
A TRUE/FALSE value to indicate if the output should be reshaped to be easier to interpret for the user, some information is missing from the multivariate models for an easy interpretation. |
stepWeight |
A vector of values (of length equal to the number of iterations) indicating the weight used to multiply the update (delta) for variance components at each iteration. If NULL the 1st iteration will be multiplied by 0.5, the 2nd by 0.7, and the rest by 0.9. This argument can help to avoid that variance components go outside the parameter space in the initial iterations which doesn't happen very often with the NR method but it can be detected by looking at the behavior of the likelihood. In that case you may want to give a smaller weight to the initial 8-10 iterations. |
emWeight |
A vector of values (of length equal to the number of iterations) indicating with values between 0 and 1 the weight assigned to the EM information matrix. And the values 1 - emWeight will be applied to the NR/AI information matrix to produce a joint information matrix. |
contrasts |
an optional list. See the contrasts.arg of model.matrix.default. |
Details
The use of this function requires a good understanding of mixed models. Please review the 'sommer.quick.start' vignette and pay attention to details like format of your random and fixed variables (e.g. character and factor variables have different properties when returning BLUEs or BLUPs, please see the 'sommer.changes.and.faqs' vignette).
For tutorials on how to perform different analysis with sommer please look at the vignettes by typing in the terminal:
vignette("v1.sommer.quick.start")
vignette("v2.sommer.changes.and.faqs")
vignette("v3.sommer.qg")
vignette("v4.sommer.gxe")
Citation
Type citation("sommer") to know how to cite the sommer package in your publications.
Special variance structures
can be used to specify heterogeneous variance for the "y" covariate at specific levels of the covariate "x", e.g., random=~vsr(at(Location,c("A","B")),ID) fits a variance component for ID at levels A and B of the covariate Location.
can be used to specify a diagonal covariance structure for the "y" covariate for all levels of the covariate "x", e.g., random=~vsr(dsr(Location),ID) fits a variance component for ID at all levels of the covariate Location.
can be used to specify an unstructured covariance structure for the "y" covariate for all levels of the covariate "x", e.g., random=~vsr(usr(Location),ID) fits variance and covariance components for ID at all levels of the covariate Location.
vsr(overlay(...,rlist=NULL,prefix=NULL))
can be used to specify overlay of design matrices between consecutive random effects specified, e.g., random=~vsr(overlay(male,female)) overlays (overlaps) the incidence matrices for the male and female random effects to obtain a single variance component for both effects. The 'rlist' argument is a list with each element being a numeric value that multiplies the incidence matrix to be overlayed. See overlay for details.Can be combined with vsr().
can be used to fit a random regression model using a numerical variable x that marks the trayectory for the random effect y. The leg function can be combined with the special functions dsr, usr at and csr. For example random=~vsr(leg(x,1),y) or random=~vsr(usr(leg(x,1)),y).
vsr(x,Gtc=fcm(v))
can be used to constrain fixed effects in the multi-response mixed models. This is a vector that specifies if the fixed effect is to be estimated for such trait. For example fixed=cbind(response.i, response.j)~vsr(Rowf, Gtc=fcm(c(1,0))) means that the fixed effect Rowf should only be estimated for the first response and the second should only have the intercept.
gvsr(x,y)
can be used to fit variance and covariance parameters between two or more random effects. For example, indirect genetic effect models.
spl2Da(x.coord, y.coord, at.var, at.levels))
can be used to fit a 2-dimensional spline (e.g., spatial modeling) using coordinates x.coord and y.coord (in numeric class) assuming a single variance component. The 2D spline can be fitted at specific levels using the at.var and at.levels arguments. For example random=~spl2Da(x.coord=Row,y.coord=Range,at.var=FIELD).
spl2Db(x.coord, y.coord, at.var, at.levels))
can be used to fit a 2-dimensional spline (e.g., spatial modeling) using coordinates x.coord and y.coord (in numeric class) assuming multiple variance components. The 2D spline can be fitted at specific levels using the at.var and at.levels arguments. For example random=~spl2Db(x.coord=Row,y.coord=Range,at.var=FIELD).
S3 methods
S3 methods are available for some parameter extraction such as fitted.mmer, residuals.mmer, summary.mmer, randef, coef.mmer, anova.mmer, plot.mmer, and predict.mmer to obtain adjusted means. In addition, the vpredict function (replacement of the pin function) can be used to estimate standard errors for linear combinations of variance components (e.g., ratios like h2).
Additional Functions
Additional functions for genetic analysis have been included such as relationship matrix building (A.mat, D.mat, E.mat, H.mat), build a genotypic hybrid marker matrix (build.HMM), plot of genetic maps (map.plot), and manhattan plots (manhattan). If you need to build a pedigree-based relationship matrix use the getA function from the pedigreemm package.
Bug report and contact
If you have any technical questions or suggestions please post it in https://stackoverflow.com or https://stats.stackexchange.com
If you have any bug report please go to https://github.com/covaruber/sommer or send me an email to address it asap, just make sure you have read the vignettes carefully before sending your question.
Example Datasets
The package has been equiped with several datasets to learn how to use the sommer package:
* DT_halfdiallel, DT_fulldiallel and DT_mohring datasets have examples to fit half and full diallel designs.
* DT_h2 to calculate heritability
* DT_cornhybrids and DT_technow datasets to perform genomic prediction in hybrid single crosses
* DT_wheat dataset to do genomic prediction in single crosses in species displaying only additive effects.
* DT_cpdata dataset to fit genomic prediction models within a biparental population coming from 2 highly heterozygous parents including additive, dominance and epistatic effects.
* DT_polyploid to fit genomic prediction and GWAS analysis in polyploids.
* DT_gryphon data contains an example of an animal model including pedigree information.
* DT_btdata dataset contains an animal (birds) model.
* DT_legendre simulated dataset for random regression model.
* DT_sleepstudy dataset to know how to translate lme4 models to sommer models.
* DT_ige dataset to show how to fit indirect genetic effect models.
Models Enabled
For details about the models enabled and more information about the covariance structures please check the help page of the package (sommer).
Value
If all parameters are correctly indicated the program will return a list with the following information:
Vi |
the inverse of the phenotypic variance matrix V^- = (ZGZ+R)^-1 |
P |
the projection matrix Vi - [Vi*(X*Vi*X)^-*Vi] |
sigma |
a list with the values of the variance-covariance components with one list element for each random effect. |
sigma_scaled |
a list with the values of the scaled variance-covariance components with one list element for each random effect. |
sigmaSE |
Hessian matrix containing the variance-covariance for the variance components. SE's can be obtained taking the square root of the diagonal values of the Hessian. |
Beta |
a data frame for trait BLUEs (fixed effects). |
VarBeta |
a variance-covariance matrix for trait BLUEs |
U |
a list (one element for each random effect) with a data frame for trait BLUPs. |
VarU |
a list (one element for each random effect) with the variance-covariance matrix for trait BLUPs. |
PevU |
a list (one element for each random effect) with the predicted error variance matrix for trait BLUPs. |
fitted |
Fitted values y.hat=XB |
residuals |
Residual values e = Y - XB |
AIC |
Akaike information criterion |
BIC |
Bayesian information criterion |
convergence |
a TRUE/FALSE statement indicating if the model converged. |
monitor |
The values of log-likelihood and variance-covariance components across iterations during the REML estimation. |
percChange |
The percent change of variance components across iterations. There should be one column less than the number of iterations. Calculated as percChange = ((x_i/x_i-1) - 1) * 100 where i is the ith iteration. |
dL |
The vector of first derivatives of the likelihood with respect to the ith variance-covariance component. |
dL2 |
The matrix of second derivatives of the likelihood with respect to the i.j th variance-covariance component. |
method |
The method for extimation of variance components specified by the user. |
call |
Formula for fixed, random and rcov used. |
constraints |
contraints used in the mixed models for the random effects. |
constraintsF |
contraints used in the mixed models for the fixed effects. |
data |
The dataset used in the model after removing missing records for the response variable. |
dataOriginal |
The original dataset used in the model. |
terms |
The name of terms for responses, fixed, random and residual effects in the model. |
termsN |
The number of effects associated to fixed, random and residual effects in the model. |
sigmaVector |
a vectorized version of the sigma element (variance-covariance components) to match easily the standard errors of the var-cov components stored in the element sigmaSE. |
reshapeOutput |
The value provided to the mmer function for the argument with the same name. |
Author(s)
Giovanny Covarrubias-Pazaran
References
Covarrubias-Pazaran G. Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 2016, 11(6): doi:10.1371/journal.pone.0156744
Covarrubias-Pazaran G. 2018. Software update: Moving the R package sommer to multivariate mixed models for genome-assisted prediction. doi: https://doi.org/10.1101/354639
Bernardo Rex. 2010. Breeding for quantitative traits in plants. Second edition. Stemma Press. 390 pp.
Gilmour et al. 1995. Average Information REML: An efficient algorithm for variance parameter estimation in linear mixed models. Biometrics 51(4):1440-1450.
Kang et al. 2008. Efficient control of population structure in model organism association mapping. Genetics 178:1709-1723.
Lee, D.-J., Durban, M., and Eilers, P.H.C. (2013). Efficient two-dimensional smoothing with P-spline ANOVA mixed models and nested bases. Computational Statistics and Data Analysis, 61, 22 - 37.
Lee et al. 2015. MTG2: An efficient algorithm for multivariate linear mixed model analysis based on genomic information. Cold Spring Harbor. doi: http://dx.doi.org/10.1101/027201.
Maier et al. 2015. Joint analysis of psychiatric disorders increases accuracy of risk prediction for schizophrenia, bipolar disorder, and major depressive disorder. Am J Hum Genet; 96(2):283-294.
Rodriguez-Alvarez, Maria Xose, et al. Correcting for spatial heterogeneity in plant breeding experiments with P-splines. Spatial Statistics 23 (2018): 52-71.
Searle. 1993. Applying the EM algorithm to calculating ML and REML estimates of variance components. Paper invited for the 1993 American Statistical Association Meeting, San Francisco.
Yu et al. 2006. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Genetics 38:203-208.
Tunnicliffe W. 1989. On the use of marginal likelihood in time series model estimation. JRSS 51(1):15-27.
Zhang et al. 2010. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 42:355-360.
Examples
####=========================================####
#### For CRAN time limitations most lines in the
#### examples are silenced with one '#' mark,
#### remove them and run the examples
####=========================================####
####=========================================####
#### EXAMPLES
#### Different models with sommer
####=========================================####
data(DT_example, package="enhancer")
DT <- DT_example
head(DT)
####=========================================####
#### Univariate homogeneous variance models ####
####=========================================####
## Compound simmetry (CS) model
ans1 <- mmer(Yield~Env,
random= ~ Name + Env:Name,
rcov= ~ units,
data=DT)
summary(ans1)
####===========================================####
#### Univariate heterogeneous variance models ####
####===========================================####
## Compound simmetry (CS) + Diagonal (DIAG) model
ans2 <- mmer(Yield~Env,
random= ~Name + vsr(dsr(Env),Name),
rcov= ~ vsr(dsr(Env),units),
data=DT)
summary(ans2)
####===========================================####
#### Univariate unstructured variance models ####
####===========================================####
ans3 <- mmer(Yield~Env,
random=~ vsr(usr(Env),Name),
rcov=~vsr(dsr(Env),units),
data=DT)
summary(ans3)
####==========================================####
#### Multivariate homogeneous variance models ####
####==========================================####
## Multivariate Compound simmetry (CS) model
DT$EnvName <- paste(DT$Env,DT$Name)
ans4 <- mmer(cbind(Yield, Weight) ~ Env,
random= ~ vsr(Name, Gtc = unsm(2)) + vsr(EnvName,Gtc = unsm(2)),
rcov= ~ vsr(units, Gtc = unsm(2)),
data=DT)
summary(ans4)
####=============================================####
#### Multivariate heterogeneous variance models ####
####=============================================####
## Multivariate Compound simmetry (CS) + Diagonal (DIAG) model
ans5 <- mmer(cbind(Yield, Weight) ~ Env,
random= ~ vsr(Name, Gtc = unsm(2)) + vsr(dsr(Env),Name, Gtc = unsm(2)),
rcov= ~ vsr(dsr(Env),units, Gtc = unsm(2)),
data=DT)
summary(ans5)
####===========================================####
#### Multivariate unstructured variance models ####
####===========================================####
ans6 <- mmer(cbind(Yield, Weight) ~ Env,
random= ~ vsr(usr(Env),Name, Gtc = unsm(2)),
rcov= ~ vsr(dsr(Env),units, Gtc = unsm(2)),
data=DT)
summary(ans6)
####=========================================####
####=========================================####
#### EXAMPLE SET 2
#### 2 variance components
#### one random effect with variance covariance structure
####=========================================####
####=========================================####
data("DT_cpdata", package="enhancer")
DT <- DT_cpdata
GT <- GT_cpdata
MP <- MP_cpdata
head(DT)
GT[1:4,1:4]
#### create the variance-covariance matrix
A <- A.mat(GT)
#### look at the data and fit the model
mix1 <- mmer(Yield~1,
random=~vsr(id, Gu=A) + Rowf,
rcov=~units,
data=DT)
summary(mix1)$varcomp
#### multi trait example
mix2 <- mmer(cbind(Yield,color)~1,
random = ~ vsr(id, Gu=A, Gtc = unsm(2)) + # unstructured at trait level
vsr(Rowf, Gtc=diag(2)) + # diagonal structure at trait level
vsr(Colf, Gtc=diag(2)), # diagonal structure at trait level
rcov = ~ vsr(units, Gtc = unsm(2)), # unstructured at trait level
data=DT)
summary(mix2)
mixed model equations solver
Description
The mmes function uses either the direct inversion or the Henderson mixed
model equations algorithms coded in C++ using the Armadillo library to optimize matrix
operations. For problems of the type c > r (more coefficients to estimate than records available),
the direct inversion algorithm is faster (using the argument henderson=FALSE; default).
For more records than coefficients set henderson=TRUE and make sure that you provide the
relationship matrix as an inverse (see vsm() function for details).
Usage
mmes(fixed, random, rcov, data, W, nIters=50, tolParConvLL = 1e-04,
tolParConvNorm = 1e-04, tolParInv = 1e-06, naMethodX="exclude",
naMethodY="exclude", returnParam=FALSE, dateWarning=TRUE,
verbose=TRUE,addScaleParam=NULL, stepWeight=NULL, emWeight=NULL,
contrasts=NULL, getPEV=TRUE, henderson=FALSE)
Arguments
fixed |
A formula specifying the response variable(s) and fixed effects, i.e: response ~ covariate |
random |
A formula specifying the name of the random effects, e.g., random= ~ genotype + year. Useful functions can be used to fit heterogeneous variances and other special models (see 'Special Functions' in the Details section for more information):
** ** ** |
rcov |
A formula specifying the name of the error term, e.g., rcov= ~ units. Special heterogeneous and special variance models and constraints for the residual part are the same used on the random term but the name of the random effect is always "units" which can be thought as a column with as many levels as rows in the data, e.g., rcov=~vsm(dsm(covariate),ism(units)) When fitting structures at the level of residuals please make sure that your data is sorted based on the factors defining the structure. For example, for rcov= ~ vsm(dsm(xx), ism(units)) sort the datatset by the variable xx. |
data |
A data frame containing the variables specified in the formulas for response, fixed, and random effects. |
W |
Weights matrix (e.g., when covariance among plots exist). Internally W is squared and inverted as Wsi = solve(chol(W)), then the residual matrix is calculated as R = Wsi*O*Wsi.t(), where * is the matrix product, and O is the original residual matrix. |
nIters |
Maximum number of iterations allowed. |
tolParConvLL |
Convergence criteria based in the change of log-likelihood between iteration i and i-1. |
tolParConvNorm |
When using the Henderson method this argument is the convergence criteria based in the norm proposed by Jensen, Madsen and Thompson (1997): e1 = || InfMatInv.diag()/sqrt(N) * dLu || where InfMatInv.diag() is the diagonal of the inverse of the information matrix, N is the total number of variance components, and dLu is the vector of first derivatives. |
tolParInv |
Tolerance parameter for matrix inverse used when singularities are encountered in the estimation procedure. By default the value is 1e-06. This parameter should be fairly small because the it is used to bend matrices like the information matrix in the henderson algorithm or the coefficient matrix when it is not positive-definite. |
naMethodX |
One of the two possible values; "include" or "exclude". If "include" is selected then the function will impute the X matrices for fixed effects with the median value. If "exclude" is selected it will get rid of all rows with missing values for the X (fixed) covariates. The default is "exclude". The "include" option should be used carefully. |
naMethodY |
One of the three possible values; "include", "include2" or "exclude" (default) to treat the observations in response variable to be used in the estimation of variance components. The first option "include" will impute the response variables for all rows with the median value, whereas "include2" imputes the responses only for rows where there is observation(s) for at least one of the responses (only available in the multi-response models). If "exclude" is selected (default) it will get rid of rows in response(s) where missing values are present for at least one of the responses. |
returnParam |
A TRUE/FALSE value to indicate if the program should return the parameters to be used for fitting the model instead of fitting the model. |
dateWarning |
A TRUE/FALSE value to indicate if the program should warn you when is time to update the sommer package. |
verbose |
A TRUE/FALSE value to indicate if the program should return the progress of the iterative algorithm. |
addScaleParam |
additional scale parameters for the thetaF matrix when using Henderson method (henderson=TRUE). |
stepWeight |
A vector of values (of length equal to the number of iterations) indicating the weight used to multiply the update (delta) for variance components at each iteration. If NULL the 1st iteration will be multiplied by 0.5, the 2nd by 0.7, and the rest by 0.9. This argument can help to avoid that variance components go outside the parameter space in the initial iterations which happens very often with the AI method but it can be detected by looking at the behavior of the likelihood. In that case you may want to give a smaller weight. |
emWeight |
A vector of values (of length equal to the number of iterations) indicating with values between 0 and 1 the weight assigned to the EM information matrix. And the values 1 - emWeight will be applied to the AI information matrix to produce a joint information matrix. By default the function gives a weight to the EM algorithm of a logarithmic decrease rate using the following code:
|
contrasts |
an optional list. See the contrasts.arg of model.matrix.default. |
getPEV |
a logical value indicating if PEV should be returned when the direct inversion algorithm is used. It does not apply when henderson argument is TRUE. |
henderson |
a logical value indicating if the solving algorithm should be direct inversion (henderson is FALSE) or Henderson's method (henderson is TRUE). Default is direct inversion. |
Details
The use of this function requires a good understanding of mixed models. Please review the 'sommer.quick.start' vignette and pay attention to details like format of your random and fixed variables (e.g. character and factor variables have different properties when returning BLUEs or BLUPs).
For tutorials on how to perform different analysis with sommer please look at the vignettes by typing in the terminal:
vignette("v1.sommer.quick.start")
vignette("v2.sommer.changes.and.faqs")
vignette("v3.sommer.qg")
vignette("v4.sommer.gxe")
Citation
Type citation("sommer") to know how to cite the sommer package in your publications.
Special variance structures
can be used to specify heterogeneous variance for the "y" covariate at specific levels of the covariate "x", e.g., random=~vsm(at(Location,c("A","B")),ism(ID)) fits a variance component for ID at levels A and B of the covariate Location.
can be used to specify a diagonal covariance structure for the "y" covariate for all levels of the covariate "x", e.g., random=~vsm(dsm(Location),ism(ID)) fits a variance component for ID at all levels of the covariate Location.
can be used to specify an unstructured covariance structure for the "y" covariate for all levels of the covariate "x", e.g., random=~vsm(usm(Location),ism(ID)) fits variance and covariance components for ID at all levels of the covariate Location.
vsm(usm(rrm(x,y,z,nPC)),ism(y))
can be used to specify an unstructured covariance structure for the "y" effect for all levels of the covariate "x", and a response variable "z", e.g., random=~vsm(rrm(Location,ID,response, nPC=2),ism(ID)) fits a reduced-rank factor analytic covariance for ID at 2 principal components of the covariate Location.
vsm(ism(overlay(...,rlist=NULL,prefix=NULL)))
can be used to specify overlay of design matrices between consecutive random
effects specified, e.g., random=~vsm(ism(overlay(male,female))) overlays
(overlaps) the incidence matrices for the male and female random effects to obtain
a single variance component for both effects. The 'rlist' argument is a list with
each element being a numeric value that multiplies the incidence matrix to be
overlayed. See overlay for details.Can be combined with vsm().
can be used to create a reduced model matrix of an effect (x) assumed to be a
linear function of some feature matrix (M), e.g., random=~vsm(ism(redmm(x,M)))
creates an incidence matrix from a very large set of features (M) that belong
to the levels of x to create a reduced model matrix. See redmm
for details.Can be combined with vsm().
can be used to fit a random regression model using a numerical variable x
that marks the trayectory for the random effect y. The leg function can
be combined with the special functions dsm, usm at and csm.
For example random=~vsm(leg(x,1),ism(y)) or random=~vsm(usm(leg(x,1)),ism(y)).
spl2Dc(x.coord, y.coord, at.var, at.levels))
can be used to fit a 2-dimensional spline (e.g., spatial modeling) using
coordinates x.coord and y.coord (in numeric class) assuming
multiple variance components. The 2D spline can be fitted at specific
levels using the at.var and at.levels arguments. For example
random=~spl2Dc(x.coord=Row,y.coord=Range,at.var=FIELD).
Covariance between random effects
covm( vsm(ism(ran1)), vsm(ism(ran2)) )
can be used to specify covariance between two different random effects, e.g., random=~covm( vsm(ism(x1)), vsm(ism(x2)) ) where two random effects in their own vsm() structure are encapsulated. Only applies for simple random effects.
S3 methods
S3 methods are available for some parameter extraction such as
fitted.mmes, residuals.mmes, summary.mmes,
randef, coef.mmes, anova.mmes,
plot.mmes, and predict.mmes to obtain adjusted means.
In addition, the vpredict function (replacement of the pin
function) can be used to estimate standard errors for linear combinations of
variance components (e.g., ratios like h2). The r2 function calculates
reliability.
Additional Functions
Additional functions for genetic analysis have been included such as relationship
matrix building (A.mat, D.mat, E.mat,
H.mat), build a genotypic hybrid marker matrix (build.HMM),
plot of genetic maps (map.plot), and manhattan plots
(manhattan). If you need to build a pedigree-based relationship
matrix use the getA function from the pedigreemm package.
Bug report and contact
If you have any technical questions or suggestions please post it in https://stackoverflow.com or https://stats.stackexchange.com
If you have any bug report please go to https://github.com/covaruber/sommer or send me an email to address it asap, just make sure you have read the vignettes carefully before sending your question.
Example Datasets
The package has been equiped with several datasets to learn how to use the sommer package:
* DT_halfdiallel, DT_fulldiallel and DT_mohring
datasets have examples to fit half and full diallel designs.
* DT_h2 to calculate heritability
* DT_cornhybrids and DT_technow datasets to perform
genomic prediction in hybrid single crosses
* DT_wheat dataset to do genomic prediction in single crosses in
species displaying only additive effects.
* DT_cpdata dataset to fit genomic prediction models within a
biparental population coming from 2 highly heterozygous parents including additive,
dominance and epistatic effects.
* DT_polyploid to fit genomic prediction and GWAS analysis in polyploids.
* DT_gryphon data contains an example of an animal model including
pedigree information.
* DT_btdata dataset contains an animal (birds) model.
* DT_legendre simulated dataset for random regression model.
* DT_sleepstudy dataset to know how to translate lme4 models to
sommer models.
* DT_ige dataset to show how to fit indirect genetic effect models.
Models Enabled
For details about the models enabled and more information about the covariance
structures please check the help page of the package (sommer).
Value
If all parameters are correctly indicated the program will return a list with the following information:
data |
the dataset used in the model fitting. |
Dtable |
the table to be used for the predict function to help the program recognize the factors available. |
llik |
the vector of log-likelihoods across iterations |
b |
the vector of fixed effect. |
u |
the vector of random effect. |
bu |
the vector of fixed and random effects together. |
Ci |
the inverse of the coefficient matrix. |
Ci_11 |
the inverse of the coefficient matrix pertaining to the fixed effects. |
theta |
a list of estimated variance covariance matrices. Each element of the list corresponds to the different random and residual components |
theta_se |
inverse of the information matrix. |
InfMat |
information matrix. |
monitor |
The values of the variance-covariance components across iterations during the REML estimation. |
AIC |
Akaike information criterion |
BIC |
Bayesian information criterion |
convergence |
a TRUE/FALSE statement indicating if the model converged. |
partitions |
a list where each element contains a matrix indicating where each random effect starts and ends. |
partitionsX |
a list where each element contains a matrix indicating where each fixed effect starts and ends. |
percDelta |
the matrix of percentage change in deltas (see tolParConvNorm argument). |
normMonitor |
the matrix of the three norms calculated (see tolParConvNorm argument). |
toBoundary |
the matrix of variance components that were forced to the boundary across iterations. |
Cchol |
the Cholesky decomposition of the coefficient matrix. |
y |
the response vector. |
W |
the column binded matrix W = [X Z] |
uList |
a list containing the BLUPs in data frame format where rows are levels of the random effects and column the different factors at which the random effect is fitted. This is specially useful for diagonal and unstructured models. |
uPevList |
a list containing the BLUPs in data frame format where rows are levels of the random effects and column the different factors at which the random effect is fitted. This is specially useful for diagonal and unstructured models. |
args |
the fixed, random and residual formulas from the mmes model. |
constraints |
The vector of constraints. |
Author(s)
Coded by Giovanny Covarrubias-Pazaran with contributions of Christelle Fernandez Camacho to the henderson algorithm.
References
Covarrubias-Pazaran G. Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 2016, 11(6): doi:10.1371/journal.pone.0156744
Jensen, J., Mantysaari, E. A., Madsen, P., and Thompson, R. (1997). Residual maximum likelihood estimation of (co) variance components in multivariate mixed linear models using average information. Journal of the Indian Society of Agricultural Statistics, 49, 215-236.
Sanderson, C., & Curtin, R. (2025). Armadillo: An Efficient Framework for Numerical Linear Algebra. arXiv preprint arXiv:2502.03000.
Gilmour et al. 1995. Average Information REML: An efficient algorithm for variance parameter estimation in linear mixed models. Biometrics 51(4):1440-1450.
Examples
####=========================================####
#### For CRAN time limitations most lines in the
#### examples are silenced with one '#' mark,
#### remove them and run the examples
####=========================================####
data(DT_example, package="enhancer")
DT <- DT_example
head(DT)
####=========================================####
#### Univariate homogeneous variance models ####
####=========================================####
## Compound simmetry (CS) model
ans1 <- mmes(Yield~Env,
random= ~ Name + Env:Name,
rcov= ~ units,
data=DT)
summary(ans1)
####===========================================####
#### Univariate heterogeneous variance models ####
####===========================================####
DT=DT[with(DT, order(Env)), ]
## Compound simmetry (CS) + Diagonal (DIAG) model
ans2 <- mmes(Yield~Env,
random= ~Name + vsm(dsm(Env),ism(Name)),
rcov= ~ vsm(dsm(Env),ism(units)),
data=DT)
summary(ans2)
####===========================================####
#### Univariate unstructured variance models ####
####===========================================####
ans3 <- mmes(Yield~Env,
random=~ vsm(usm(Env),ism(Name)),
rcov=~vsm(dsm(Env),ism(units)),
data=DT)
summary(ans3)
plot form a LMM plot with mmes
Description
plot method for class "mmes".
Usage
## S3 method for class 'mmes'
plot(x,stnd=TRUE, ...)
Arguments
x |
an object of class |
stnd |
argument for ploting the residuals to know if they should be standarized. |
... |
Further arguments to be passed |
Value
vector of plot
Author(s)
Giovanny Covarrubias covarrubiasp@wisc.edu
See Also
Examples
data(DT_yatesoats, package="enhancer")
DT <- DT_yatesoats
head(DT)
m3 <- mmes(fixed=Y ~ V + N + V:N,
random = ~ B + B:MP,
rcov=~units,
data = DT)
plot(m3)
plot the change of VC across iterations
Description
plot for monitoring.
Usage
pmonitor(object, ...)
Arguments
object |
model object of class |
... |
Further arguments to be passed to the plot function. |
Value
vector of plot
Author(s)
Giovanny Covarrubias
See Also
Examples
data(DT_yatesoats, package="enhancer")
DT <- DT_yatesoats
head(DT)
m3 <- mmes(fixed=Y ~ V + N + V:N,
random = ~ B + B:MP,
rcov=~units,
data = DT)
pmonitor(m3)
Predict form of a LMM fitted with mmes
Description
predict method for class "mmes".
Usage
## S3 method for class 'mmes'
predict(object, Dtable=NULL, D, ...)
Arguments
object |
a mixed model of class |
Dtable |
a table specifying the terms to be included or averaged. An "include" term means that the model matrices for that fixed or random effect is filled with 1's for the positions where column names and row names match. An "include and average" term means that the model matrices for that fixed or random effect is filled with 1/#1's in that row. An "average" term alone means that all rows for such fixed or random effect will be filled with 1/#levels in the effect. If a term is not considered "include" or "average" is then totally ignored in the BLUP and SE calculation. The default rule to invoke when the user doesn't provide the Dtable is to include and average all terms that match the argument D. |
D |
a character string specifying the variable used to extract levels for the rows of the D matrix and its construction. Alternatively, the D matrix (of class dgCMatrix) specifying the matrix to be used for the predictions directly. |
... |
Further arguments to be passed. |
Details
This function allows to produce predictions specifying those variables that define the margins of the hypertable to be predicted (argument D). Predictions are obtained for each combination of values of the specified variables that is present in the data set used to fit the model. See vignettes for more details.
For predicted values the pertinent design matrices X and Z together with BLUEs (b) and BLUPs (u) are multiplied and added together.
predicted.value equal Xb + Zu.1 + ... + Zu.n
For computing standard errors for predictions the parts of the coefficient matrix:
C11 equal (X.t() V.inv() X).inv()
C12 equal 0 - [(X.t() V.inv() X).inv() X.t() V.inv() G Z]
C22 equal PEV equal G - [Z.t() G[V.inv() - (V.inv() X X.t() V.inv() X V.inv() X)]G Z.t()]
In practive C equals ( W.t() V.inv() W ).inv()
when both fixed and random effects are present in the inclusion set. If only fixed and random effects are included, only the respective terms from the SE for fixed or random effects are calculated.
Value
pvals |
the table of predictions according to the specified arguments. |
vcov |
the variance covariance for the predictions. |
D |
the model matrix for predictions as defined in Welham et al.(2004). |
Dtable |
the table specifying the terms to include and terms to be averaged. |
Author(s)
Giovanny Covarrubias-Pazaran
References
Welham, S., Cullis, B., Gogel, B., Gilmour, A., and Thompson, R. (2004). Prediction in linear mixed models. Australian and New Zealand Journal of Statistics, 46, 325 - 347.
See Also
Examples
data(DT_yatesoats, package="enhancer")
DT <- DT_yatesoats
m3 <- mmes(fixed=Y ~ V + N + V:N ,
random = ~ B + B:MP,
rcov=~units,
data = DT)
#############################
## predict means for nitrogen
#############################
Dt <- m3$Dtable; Dt
# first fixed effect just average
Dt[1,"average"] = TRUE
# second fixed effect include
Dt[2,"include"] = TRUE
# third fixed effect include and average
Dt[3,"include"] = TRUE
Dt[3,"average"] = TRUE
Dt
pp=predict(object=m3, Dtable=Dt, D="N")
pp$pvals
#############################
## predict means for variety
#############################
Dt <- m3$Dtable; Dt
# first fixed effect include
Dt[1,"include"] = TRUE
# second fixed effect just average
Dt[2,"average"] = TRUE
# third fixed effect include and average
Dt[3,"include"] = TRUE
Dt[3,"average"] = TRUE
Dt
pp=predict(object=m3, Dtable=Dt, D="V")
pp$pvals
#############################
## predict means for nitrogen:variety
#############################
# prediction matrix D based on (equivalent to classify in asreml)
Dt <- m3$Dtable; Dt
# first fixed effect include and average
Dt[1,"include"] = TRUE
Dt[1,"average"] = TRUE
# second fixed effect include and average
Dt[2,"include"] = TRUE
Dt[2,"average"] = TRUE
# third fixed effect include and average
Dt[3,"include"] = TRUE
Dt[3,"average"] = TRUE
Dt
pp=predict(object=m3, Dtable=Dt, D="N:V")
pp$pvals
Reliability
Description
Calculates the reliability of BLUPs in a sommer model.
Usage
r2(object, object2=NULL)
Arguments
object |
Model fitted with the mmes function. |
object2 |
An optional model identical to object in the first argument but fitted with the argument returnParam set to TRUE to access the relationship matrices from the fitted model. |
Details
The reliability method calculated is the classical animal model: R2=(G-PEV)/G
Value
- result
a list with as many elements as random effects fitted containing reliabilities for individual BLUPs.
References
Mrode, R. A. (2014). Linear models for the prediction of animal breeding values. Cabi.
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
See Also
mmes – the core function of the package
Examples
####=========================================####
#### Example population
####=========================================####
data(DT_example, package="enhancer")
DT <- DT_example
head(DT)
ans1 <- mmes(Yield~Env,
random= ~ Name + Env:Name,
rcov= ~ units,
data=DT)
rel=r2(ans1)
extracting random effects
Description
This function is extracts the random effects from a mixed model fitted by mmer.
Usage
randef(object)
Arguments
object |
an mmer object |
Value
- $randef
a list structure with the random effects or BLUPs.
Examples
# randef(model)
Residuals form a GLMM fitted with mmes
Description
residuals method for class "mmes".
Usage
## S3 method for class 'mmes'
residuals(object, ...)
Arguments
object |
an object of class |
... |
Further arguments to be passed |
Value
vector of residuals of the form e = y - Xb - Zu, the so called conditional residuals.
Author(s)
Giovanny Covarrubias
See Also
Solving Mixed Model Equations in R

Description
Sommer is a structural multivariate-univariate linear mixed model solver for multiple random effects allowing the specification and/or estimation of variance covariance structures. REML estimates can be obtained using two major methods
Direct-Inversion (Newton-Raphson and Average Information)
Henderson's mixed model equations (Average Information)
The algorithms are coded in C++ using the Armadillo library to optimize dense matrix operations common in genomic models. Sommer was designed to include complex covariance structures, e.g., unstructured, reduced-rank, diagonal. And also to model relationships between levels of a random effect, e.g., additive, dominance and epistatic relationship structures.
The direct inversion algorithm available in the mmes function
(argument henderson=FALSE) can
deal well with small and medium-size data sets (< 10,000 observations/records for
average computers given the computational burden carried by the direct-inversion
algorithms) since it works in the c > r problem and inverts an r x r matrix
(being r the number of records and c the number of coefficients). On the other hand,
the Henderson algorithm in the mmes function (argument henderson=TRUE)
can deal with greater number of records (r>250K) as long as the
number of coefficients to estimate is < 10,000 coefficients (c) since it works in
the r > c problem and inverts a c x c matrix (being c the number of coefficients).
The predict.mmes function can be used to obtain adjusted means.
This package returns variance-covariance components, BLUPs, BLUEs, residuals,
fitted values, variances-covariances for fixed and random effects, etc.
Functions for genetic analysis
The package provides kernels to estimate additive (A.mat), dominance
(D.mat), epistatic (E.mat), single-step (H.mat)
relationship matrices for diploid and polyploid organisms. It also provides flexibility
to fit other genetic models such as full and half diallel models and random regression models.
A good converter from letter code to numeric format is implemented in the function
atcg1234, which supports higher ploidy levels than diploid.
Additional functions for genetic analysis have been included such as build a
genotypic hybrid marker matrix (build.HMM), plot of genetic maps
(map.plot), creation of manhattan plots (manhattan).
If you need to use pedigree you need to convert your pedigree into a relationship
matrix (use the 'getA' function from the pedigreemm package).
Functions for statistical analysis and S3 methods
The vpredict function can be used to estimate standard errors for linear combinations of variance components (e.g. ratios like h2). The r2 function calculates reliability. S3 methods are available for some parameter extraction such as:
Functions for trial analysis
Recently, spatial modeling has been added added to sommer using the two-dimensional
spline (spl2Dc).
Keeping sommer updated
The sommer package is updated on CRAN every 4-months due to CRAN policies but you can find the latest source at https://github.com/covaruber/sommer. This can be easily installed typing the following in the R console:
library(devtools)
install_github("covaruber/sommer")
This is recommended if you reported a bug, was fixed and was immediately pushed to GitHub but not in CRAN until the next update.
Tutorials
For tutorials on how to perform different analysis with sommer please look at the vignettes by typing in the terminal:
vignette("sommer.qg")
vignette("sommer.gxe")
vignette("sommer.vs.lme4")
vignette("sommer.spatial")
Getting started
The package has been equiped with several datasets to learn how to use the sommer package (and almost to learn all sort of quantitative genetic analysis):
* DT_halfdiallel, DT_fulldiallel and DT_mohring datasets have examples to fit half and full diallel designs.
* DT_h2 to calculate heritability
* DT_cornhybrids and DT_technow datasets to perform genomic prediction in hybrid single crosses
* DT_wheat dataset to do genomic prediction in single crosses in species displaying only additive effects.
* DT_cpdata dataset to fit genomic prediction models within a biparental population coming from 2 highly heterozygous parents including additive, dominance and epistatic effects.
* DT_polyploid to fit genomic prediction and GWAS analysis in polyploids.
* DT_gryphon data contains an example of an animal model including pedigree information.
* DT_btdata dataset contains an animal (birds) model.
* DT_legendre simulated dataset for random regression model.
* DT_sleepstudy dataset to know how to translate lme4 models to sommer models.
Differences of sommer >= 4.4.1 with previous versions
Since version 4.4.1, I have unified the use of the two different solving algorithms
into the mmes function by just using the new argument henderson which by default
is set to FALSE. Other than that the rest is the same with the addition that now
the identity terms needs to be encapsulated in the ism function. In
addition, now the multi-trait models need to be fitted in the long format. This are
few but major changes to the way sommer models are fitted.
Differences of sommer >= 4.1.7 with previous versions
Since version 4.1.7 I have introduced the mmes-based average information function 'mmec' which is much faster when dealing with the r > c problem (more records than coefficients to estimate). This introduces its own covariance structure functons such as vsc(), usc(), dsc(), atc(), csc(). Please give it a try, although is in early phase of development.
Differences of sommer >= 3.7.0 with previous versions
Since version 3.7 I have completly redefined the specification of the variance-covariance structures to provide more flexibility to the user. This has particularly helped the residual covariance structures and the easier combination of custom random effects and overlay models. I think that although this will bring some uncomfortable situations at the beggining, in the long term this will help users to fit better models. In esence, I have abandoned the asreml formulation (not the structures available) given it's limitations to combine some of the sommer structures but all covariance structures can now be fitted using the 'vsm' functions.
Differences of sommer >= 3.0.0 with previous versions
Since version 3.0 I have decided to focus in developing the multivariate solver and for doing this I have decided to remove the M argument (for GWAS analysis) from the mmes function and move it to it's own function GWAS.
Before the mmes solver had implemented the usm(trait), diag(trait), at(trait) asreml formulation for multivariate models that allow to specify the structure of the trait in multivariate models. Therefore the MVM argument was no longer needed. After version 3.7 now the multi-trait structures can be specified in the Gt and Gtc arguments of the vsm function.
The Average Information algorithm had been removed in the past from the package because of its instability to deal with very complex models without good initial values. Now after 3.7 I have brought it back after I noticed that starting with NR the first three iterations gives enough flexibility to the AI algorithm.
Keep in mind that sommer uses direct inversion (DI) algorithm which can be very slow for datasets with many observations (big 'n'). The package is focused in problems of the type p > n (more random effect(s) levels than observations) and models with dense covariance structures. For example, for experiment with dense covariance structures with low-replication (i.e. 2000 records from 1000 individuals replicated twice with a covariance structure of 1000x1000) sommer will be faster than MME-based software. Also for genomic problems with large number of random effect levels, i.e. 300 individuals (n) with 100,000 genetic markers (p). On the other hand, for highly replicated trials with small covariance structures or n > p (i.e. 2000 records from 200 individuals replicated 10 times with covariance structure of 200x200) asreml or other MME-based algorithms will be much faster and I recommend you to use that software.
Models Enabled
The core of the package are the mmes (formula-based) function which
solve the mixed model equations. The functions are an interface to call the 'NR'
Direct-Inversion Newton-Raphson, 'AI' Direct-Inversion Average Information or
the mmes-based Average Information (Tunnicliffe 1989; Gilmour et al. 1995;
Lee et al. 2016). Since version 2.0 sommer can handle multivariate models.
Following Maier et al. (2015), the multivariate (and by extension the univariate)
mixed model implemented has the form:
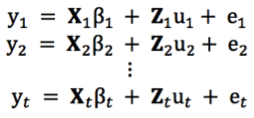
where y_i is a vector of trait phenotypes, \beta_i is a vector of fixed effects, u_i is a vector of random effects for individuals and e_i are residuals for trait i (i = 1,..., t). The random effects (u_1 ... u_i and e_i) are assumed to be normally distributed with mean zero. X and Z are incidence matrices for fixed and random effects respectively. The distribution of the multivariate response and the phenotypic variance covariance (V) are:
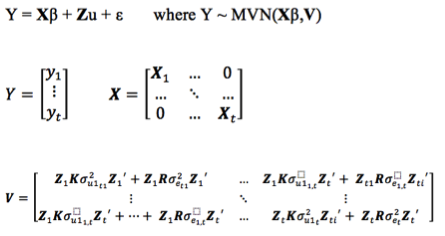
where K is the relationship or covariance matrix for the kth random effect (u=1,...,k), and R=I is an identity matrix for the residual term. The terms \sigma^2_{g_{i}} and \sigma^2_{\epsilon_{i}} denote the genetic (or any of the kth random terms) and residual variance of trait i, respectively and \sigma_{g_{_{ij}}} and \sigma_{\epsilon_{_{ij}}} the genetic (or any of the kth random terms) and residual covariance between traits i and j (i=1,...,t, and j=1,...,t). The algorithm implemented optimizes the log likelihood:
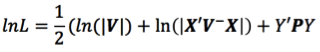
where || is the determinant of a matrix. And the REML estimates are updated using a Newton optimization algorithm of the form:
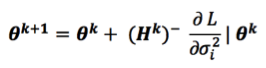
Where, theta is the vector of variance components for random effects and covariance components among traits, H^-1 is the inverse of the Hessian matrix of second derivatives for the kth cycle, dL/dsigma^2_i is the vector of first derivatives of the likelihood with respect to the variance-covariance components. The Eigen decomposition of the relationship matrix proposed by Lee and Van Der Werf (2016) was included in the Newton-Raphson algorithm to improve time efficiency. Additionally, the popular vpredict function to estimate standard errors for linear combinations of variance components (i.e. heritabilities and genetic correlations) was added to the package as well.
Bug report and contact
If you have any questions or suggestions please post it in https://stackoverflow.com or https://stats.stackexchange.com
I'll be glad to help or answer any question. I have spent a valuable amount of time developing this package. Please cite this package in your publication. Type 'citation("sommer")' to know how to cite it.
Author(s)
Giovanny Covarrubias-Pazaran
References
Covarrubias-Pazaran G. 2016. Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Covarrubias-Pazaran G. 2018. Software update: Moving the R package sommer to multivariate mixed models for genome-assisted prediction. doi: https://doi.org/10.1101/354639
Sanderson, C., & Curtin, R. (2025). Armadillo: An Efficient Framework for Numerical Linear Algebra. arXiv preprint arXiv:2502.03000.
Bernardo Rex. 2010. Breeding for quantitative traits in plants. Second edition. Stemma Press. 390 pp.
Gilmour et al. 1995. Average Information REML: An efficient algorithm for variance parameter estimation in linear mixed models. Biometrics 51(4):1440-1450.
Henderson C.R. 1975. Best Linear Unbiased Estimation and Prediction under a Selection Model. Biometrics vol. 31(2):423-447.
Kang et al. 2008. Efficient control of population structure in model organism association mapping. Genetics 178:1709-1723.
Lee et al. 2015. MTG2: An efficient algorithm for multivariate linear mixed model analysis based on genomic information. Cold Spring Harbor. doi: http://dx.doi.org/10.1101/027201.
Maier et al. 2015. Joint analysis of psychiatric disorders increases accuracy of risk prediction for schizophrenia, bipolar disorder, and major depressive disorder. Am J Hum Genet; 96(2):283-294.
Searle. 1993. Applying the EM algorithm to calculating ML and REML estimates of variance components. Paper invited for the 1993 American Statistical Association Meeting, San Francisco.
Yu et al. 2006. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Genetics 38:203-208.
Tunnicliffe W. 1989. On the use of marginal likelihood in time series model estimation. JRSS 51(1):15-27.
Examples
####=========================================####
#### For CRAN time limitations most lines in the
#### examples are silenced with one '#' mark,
#### remove them and run the examples
####=========================================####
####=========================================####
#### EXAMPLES
#### Different models with sommer
####=========================================####
data(DT_example, package="enhancer")
DT <- DT_example
DT=DT[with(DT, order(Env)), ]
head(DT)
####=========================================####
#### Univariate homogeneous variance models ####
####=========================================####
## Compound simmetry (CS) model
ans1 <- mmes(Yield~Env,
random= ~ Name + Env:Name,
rcov= ~ units,
data=DT)
summary(ans1)
####===========================================####
#### Univariate heterogeneous variance models ####
####===========================================####
## Compound simmetry (CS) + Diagonal (DIAG) model
ans3 <- mmes(Yield~Env,
random= ~Name + vsm(dsm(Env),ism(Name)),
rcov= ~ vsm(dsm(Env),ism(units)),
data=DT)
summary(ans3)
Two-dimensional penalised tensor-product of marginal B-Spline basis.
Description
Auxiliary function used for modelling the spatial or environmental effect as a two-dimensional penalised tensor-product (isotropic approach) based on Lee et al. (2013) and Rodriguez-Alvarez et al. (2018). This is a modified wrapper of some portions of the SpATS package to build a single incidence matrix containing all the columns from tensor products of the x and y coordinates and it fits such matrix as a single random effect. Then the heterogeneous covariances structure capabilities of sommer can be used to enhance the model fit. You may be interested in reading and citing not only sommec but also Wageningen publications if using this 2D spline methodology.
Usage
spl2Dc(x.coord,y.coord,at.var=NULL,at.levels=NULL, type="PSANOVA",
nsegments = c(10,10), penaltyord = c(2,2), degree = c(3,3),
nestorder = c(1,1), thetaC=NULL, theta=NULL, sp=FALSE)
Arguments
x.coord |
vector of coordinates on the x-axis direction (i.e. row) to use in the 2 dimensional spline. |
y.coord |
vector of coordinates on the y-axis direction (i.e. range or column) to use in the 2 dimensional spline. |
at.var |
vector of indication variable where heterogeneous variance is required (e.g., a different spl2D for each field). |
at.levels |
character vector with the names of the leves for the at term that should be used, if missing all levels are used. |
type |
one of the two methods "PSANOVA" or "SAP". See details below. |
nsegments |
numerical vector of length 2 containing the number of segments for each marginal (strictly |
penaltyord |
numerical vector of length 2 containing the penalty order for each marginal. Atomic values are also valid, being recycled. Default set to 2 (second order). Currently, only second order penalties are allowed. |
degree |
numerical vector of length 2 containing the order of the polynomial of the B-spline basis for each marginal. Atomic values are also valid, being recycled. Default set to 3 (cubic B-splines). |
nestorder |
numerical vector of length 2 containing the divisor of the number of segments ( |
thetaC |
an optional matrix for constraints in the variance components. |
theta |
an optional matrix for initial values of the variance components. |
sp |
a TRUE/FALSE statement to indicate if the VC from this structure should be multiplied by the scale parameter added in the mmes function through the addScaleParam argument in the |
Details
The following documentation is taken from the SpATS package. Please refer to this package and associated publications if you are interested in going deeper on this technique:
Within the P-spline framework, anisotropic low-rank tensor-product smoothers have become the general approach for modelling multidimensional surfaces (Eilers and Marx 2003; Wood 2006). In the original SpATS package, was proposed to model the spatial or environmental effect by means of the tensor-product of B-splines basis functions. In other words, was proposed to model the spatial trend as a smooth bivariate surface jointly defined over the the spatial coordinates. Accordingly, the current function has been designed to allow the user to specify the spatial coordinates that the spatial trend is a function of. There is no restriction about how the spatial coordinates shall be specified: these can be the longitude and latitude of the position of the plot on the field or the column and row numbers. The only restriction is that the variables defining the spatial coordinates should be numeric (in contrast to factors).
As far as estimation is concerned, we have used in this package the equivalence between P-splines and linear mixed models (Currie and Durban, 2002). Under this approach, the smoothing parameters are expressed as the ratio between variance components. Moreover, the smooth components are decomposed in two parts: one which is not penalised (and treated as fixed) and one with is penalised (and treated as random). For the two-dimensional case, the mixed model representation leads also to a very interesting decomposition of the penalised part of the bivariate surface in three different components (Lee and Durban, 2011): (a) a component that contains the smooth main effect (smooth trend) along one of the covariates that the surface is a function of (as, e.g, the x-spatial coordinate or column position of the plot in the field), (b) a component that contains the smooth main effect (smooth trend) along the other covariate (i.e., the y-spatial coordinate or row position); and (c) a smooth interaction component (sum of the linear-by-smooth interaction components and the smooth-by-smooth interaction component).
The original implementation of SpATS assumes two different smoothing parameters, i.e., one for each covariate in the smooth component. Accordingly, the same smoothing parameters are used for both, the main effects and the smooth interaction. However, this approach can be extended to deal with the ANOVA-type decomposition presented in Lee and Durban (2011). In their approach, four different smoothing parameters are considered for the smooth surface, that are in concordance with the aforementioned decomposition: (a) two smoothing parameter, one for each of the main effects; and (b) two smoothing parameter for the smooth interaction component.
It should be noted that, the computational burden associated with the estimation of the two-dimensional tensor-product smoother might be prohibitive if the dimension of the marginal bases is large. In these cases, Lee et al. (2013) propose to reduce the computational cost by using nested bases. The idea is to reduce the dimension of the marginal bases (and therefore the associated number of parameters to be estimated), but only for the smooth-by-smooth interaction component. As pointed out by the authors, this simplification can be justified by the fact that the main effects would in fact explain most of the structure (or spatial trend) presented in the data, and so a less rich representation of the smooth-by-smooth interaction component could be needed. In order to ensure that the reduced bivariate surface is in fact nested to the model including only the main effects, Lee et al. (2013) show that the number of segments used for the nested basis should be a divisor of the number of segments used in the original basis (nsegments argument). In the present function, the divisor of the number of segments is specified through the argument nestorder. For a more detailed review on this topic, see Lee (2010) and Lee et al. (2013). The "PSANOVA" approach represents an alternative method. In this case, the smooth bivariate surface (or spatial trend) is decomposed in five different components each of them depending on a single smoothing parameter (see Lee et al., 2013).
—————–
As mentioned at the beginning, the piece of documentation stated above was taken completely from the SpATS package in order to provide a deeper explanation. In practice, sommec uses some pieces of code from SpATS to build the design matrix containing all the columns from tensor products of the x and y coordinates and it fits such matrix as a single random effect. As a result the same variance component is assumed for the linear, linear by linear, linear by spline, and spline by spline interactions. This results in a less flexible approach than the one proposed by Rodriguez-Alvarez et al. (2018) but still makes a pretty good job to model the spatial variation. Use under your own risk.
References
Rodriguez-Alvarez, M.X, Boer, M.P., van Eeuwijk, F.A., and Eilers, P.H.C. (2018). SpATS: Spatial Analysis of Field Trials with Splines. R package version 1.0-9. https://CRAN.R-project.org/package=SpATS.
Rodriguez-Alvarez, M.X., et al. (2015) Fast smoothng parameter separaton n multdmensonal generalzed P-splnes: the SAP algorthm. Statistics and Computing 25.5: 941-957.
Lee, D.-J., Durban, M., and Eilers, P.H.C. (2013). Efficient two-dimensional smoothing with P-spline ANOVA mixed models and nested bases. Computational Statistics and Data Analysis, 61, 22 - 37.
Gilmour, A.R., Cullis, B.R., and Verbyla, A.P. (1997). Accounting for Natural and Extraneous Variation in the Analysis of Field Experiments. Journal of Agricultural, Biological, and Environmental Statistics, 2, 269 - 293.
See Also
Examples
## ============================ ##
## example to use spl2Dc()
## ============================ ##
data(DT_cpdata, package="enhancer")
# DT <- DT_cpdata
# GT <- GT_cpdata
# MP <- MP_cpdata
# A <- A.mat(GT)
## ============================ ##
## mimic 2 fields
## ============================ ##
# aa <- DT; bb <- DT
# aa$FIELD <- "A";bb$FIELD <- "B"
# set.seed(1234)
# aa$Yield <- aa$Yield + rnorm(length(aa$Yield),0,4)
# DT2 <- rbind(aa,bb)
# head(DT2)
# mix <- mmes(Yield~1, henderson = F,
# random=~
# vsm(dsm(FIELD),ism(Rowf)) +
# vsm(dsm(FIELD),ism(Colf)) +
# spl2Dc(Row,Col,at.var=FIELD),
# rcov=~vsm(dsm(FIELD),ism(units)),
# data=DT2)
#
# # extract spatial effects
# blup <- mix$uList$`spl2Dc(Row, Col, at.var = FIELD`
# head(blup) # 2 fields
# # recreate the incidence matrices
# xx=with(DT2, spl2Dc(Row,Col,at.var=FIELD))
# # get fitted values Zu for spatial effects and add them to the dataset
# field1 <- xx$Z$`A:all` %*% blup[,1]
# field2 <- xx$Z$`B:all` %*% blup[,2]
# DT2$spat <- field1+field2
# # plots the spatial effects
# lattice::levelplot(spat~Row*Col|FIELD, data=DT2)
Get Tensor Product Spline Mixed Model Incidence Matrices
Description
spl2Dmats gets Tensor-Product P-Spline Mixed Model Incidence Matrices
for use with sommer and its main function mmes. We thank Sue Welham for making the TPSbits package available to the community. If you're using this function for your research please cite her TPSbits package :) this is mostly a wrapper of her tpsmmb function to enable the use in sommer.
Usage
spl2Dmats(
x.coord.name,
y.coord.name,
data,
at.name,
at.levels,
nsegments=NULL,
minbound=NULL,
maxbound=NULL,
degree = c(3, 3),
penaltyord = c(2,2),
nestorder = c(1,1),
method = "Lee"
)
Arguments
x.coord.name |
A string. Gives the name of |
y.coord.name |
A string. Gives the name of |
data |
A dataframe. Holds the dataset to be used for fitting. |
at.name |
name of a variable defining if the 2D spline matrices should be created at different units (e.g., at different environments). |
at.levels |
a vector of names indicating which levels of the at.name variable should be used for fitting the 2D spline function. |
nsegments |
A list of length 2. Number of segments to split column and row ranges into, respectively (= number of internal knots + 1). If only one number is specified, that value is used in both dimensions. If not specified, (number of unique values - 1) is used in each dimension; for a grid layout (equal spacing) this gives a knot at each data value. |
minbound |
A list of length 2. The lower bound to be used for column and row dimensions respectively; default calculated as the minimum value for each dimension. |
maxbound |
A list of length 2. The upper bound to be used for column and row dimensions respectively; default calculated as the maximum value for each dimension. |
degree |
A list of length 2. The degree of polynomial spline to be used for column and row dimensions respectively; default=3. |
penaltyord |
A list of length 2. The order of differencing for column and row dimensions, respectively; default=2. |
nestorder |
A list of length 2. The order of nesting for column and row dimensions, respectively; default=1 (no nesting). A value of 2 generates a spline with half the number of segments in that dimension, etc. The number of segments in each direction must be a multiple of the order of nesting. |
method |
A string. Method for forming the penalty; default= |
Value
List of length 7 elements:
-
data= the input data frame augmented with structures required to fit tensor product splines inasreml-R. This data frame can be used to fit the TPS model.Added columns:
-
TP.col,TP.row= column and row coordinates -
TP.CxR= combined index for use with smooth x smooth term -
TP.C.nfor n=1:(diff.c) = X parts of column spline for use in random model (where diff.c is the order of column differencing) -
TP.R.nfor n=1:(diff.r) = X parts of row spline for use in random model (where diff.r is the order of row differencing) -
TP.CR.nfor n=1:((diff.c*diff.r)) = interaction between the two X parts for use in fixed model. The first variate is a constant term which should be omitted from the model when the constant (1) is present. If all elements are included in the model then the constant term should be omitted, eg.y ~ -1 + TP.CR.1 + TP.CR.2 + TP.CR.3 + TP.CR.4 + other terms... when
asreml="grp"or"sepgrp", the spline basis functions are also added into the data frame. Column numbers for each term are given in thegrplist structure.
-
-
fR= Xr1:Zc -
fC= Xr2:Zc -
fR.C= Zr:Xc1 -
R.fC= Zr:Xc2 -
fR.fC= Zc:Zr -
all= Xr1:Zc | Xr2:Zc | Zr:Xc1 | Zr:Xc2 | Zc:Zr
Examples
data("DT_cpdata", package="enhancer")
DT <- DT_cpdata
GT <- GT_cpdata
MP <- MP_cpdata
#### create the variance-covariance matrix
A <- A.mat(GT) # additive relationship matrix
M <- spl2Dmats(x.coord.name = "Col", y.coord.name = "Row", data=DT, nseg =c(14,21))
head(M$data)
# m1g <- mmes(Yield~1+TP.CR.2+TP.CR.3+TP.CR.4,
# random=~Rowf+Colf+vsm(ism(M$fC))+vsm(ism(M$fR))+
# vsm(ism(M$fC.R))+vsm(ism(M$C.fR))+vsm(ism(M$fC.fR))+
# vsm(ism(id),Gu=A),
# data=M$data, tolpar = 1e-6,
# iters=30)
#
# summary(m1g)$varcomp
summary form a GLMM fitted with mmer
Description
summary method for class "mmer".
Usage
## S3 method for class 'mmer'
summary(object, ...)
Arguments
object |
an object of class |
... |
Further arguments to be passed |
Value
vector of summary
Author(s)
Giovanny Covarrubias-Pazaran
See Also
summary form a GLMM fitted with mmes
Description
summary method for class "mmes".
Usage
## S3 method for class 'mmes'
summary(object, ...)
Arguments
object |
an object of class |
... |
Further arguments to be passed |
Value
vector of summary
Author(s)
Giovanny Covarrubias-Pazaran
See Also
Get Tensor Product Spline Mixed Model Incidence Matrices
Description
tpsmmbwrapper is a wrapper of tpsmmb function from the TPSbits package to avoid version dependencies but if you're using this function for your research please cite the TPSbits package. This function is internally used by the spl2Dmatrices function to get Tensor-Product P-Spline Mixed Model Bits
(design matrices) for use with sommer.
Usage
tpsmmbwrapper(
columncoordinates,
rowcoordinates,
data,
nsegments=NULL,
minbound=NULL,
maxbound=NULL,
degree = c(3, 3),
penaltyord = c(2, 2),
nestorder = c(1, 1),
asreml = "mbf",
eigenvalues = "include",
method = "Lee",
stub = NULL
)
Arguments
columncoordinates |
A string. Gives the name of |
rowcoordinates |
A string. Gives the name of |
data |
A dataframe. Holds the dataset to be used for fitting. |
nsegments |
A list of length 2. Number of segments to split column and row ranges into, respectively (= number of internal knots + 1). If only one number is specified, that value is used in both dimensions. If not specified, (number of unique values - 1) is used in each dimension; for a grid layout (equal spacing) this gives a knot at each data value. |
minbound |
A list of length 2. The lower bound to be used for column and row dimensions respectively; default calculated as the minimum value for each dimension. |
maxbound |
A list of length 2. The upper bound to be used for column and row dimensions respectively; default calculated as the maximum value for each dimension. |
degree |
A list of length 2. The degree of polynomial spline to be used for column and row dimensions respectively; default=3. |
penaltyord |
A list of length 2. The order of differencing for column and row dimensions, respectively; default=2. |
nestorder |
A list of length 2. The order of nesting for column and row dimensions, respectively; default=1 (no nesting). A value of 2 generates a spline with half the number of segments in that dimension, etc. The number of segments in each direction must be a multiple of the order of nesting. |
asreml |
A string. Indicates the types of structures to be generated
for use in asreml models; default
|
eigenvalues |
A string. Indicates whether eigenvalues should be
included within the Z design matrices |
method |
A string. Method for forming the penalty; default= |
stub |
A string. Stub to be attached to names in the |
Value
List of length 7, 8 or 9 (according to the asreml and
eigenvalues parameter settings).
-
data= the input data frame augmented with structures required to fit tensor product splines inasreml-R. This data frame can be used to fit the TPS model.Added columns:
-
TP.col,TP.row= column and row coordinates -
TP.CxR= combined index for use with smooth x smooth term -
TP.C.nfor n=1:(diff.c) = X parts of column spline for use in random model (where diff.c is the order of column differencing) -
TP.R.nfor n=1:(diff.r) = X parts of row spline for use in random model (where diff.r is the order of row differencing) -
TP.CR.nfor n=1:((diff.c*diff.r)) = interaction between the two X parts for use in fixed model. The first variate is a constant term which should be omitted from the model when the constant (1) is present. If all elements are included in the model then the constant term should be omitted, eg.y ~ -1 + TP.CR.1 + TP.CR.2 + TP.CR.3 + TP.CR.4 + other terms... when
asreml="grp"or"sepgrp", the spline basis functions are also added into the data frame. Column numbers for each term are given in thegrplist structure.
-
-
mbflist= list that can be used in call to asreml (so long as Z matrix data frames extracted with right names, eg BcZ<stub>.df) -
BcZ.df= mbf data frame mapping onto smooth part of column spline, last column (labelledTP.col) gives column index -
BrZ.df= mbf data frame mapping onto smooth part of row spline, last column (labelledTP.row) gives row index -
BcrZ.df= mbf data frame mapping onto smooth x smooth term, last column (labelledTP.CxR) maps onto col x row combined index -
dim= list structure, holding dimension values relating to the model:-
"diff.c"= order of differencing used in column dimension -
"nbc"= number of random basis functions in column dimension -
"nbcn"= number of nested random basis functions in column dimension used in smooth x smooth term -
"diff.r"= order of differencing used in column dimension -
"nbr"= number of random basis functions in column dimension -
"nbrn"= number of nested random basis functions in column dimension used in smooth x smooth term
-
-
trace= list of trace values for ZGZ' for the random TPspline terms, where Z is the design matrix and G is the known diagonal variance matrix derived from eigenvalues. This can be used to rescale the spline design matrix (or equivalently variance components). -
grp= list structure, only added for settingsasreml="grp",asreml="sepgrp"orasreml="own". Forasreml="grp", provides column indexes for each of the 5 random components of the 2D splines. Forasreml="sepgrp", provides column indexes for each of the X and Z component matrices for the 1D splines, plus the composite smooth x smooth interaction term. Forasreml="own", provides column indexes for the composite random model. Dimensions of the components can be derived from the values in thedimitem. The Z terms are scaled by the associated eigenvalues wheneigenvalues="include", but not wheneigenvalues="omit". -
eigen= list structure, only added for option settingeigenvalues="omit". Holds the diagonal elements of the inverse variance matrix for the terms Xc:Zr (calleddiagr), Zc:Xr (calleddiagc) and Zc:Zr (calleddiagcr).
unstructured indication matrix
Description
unsm creates a square matrix with ones in the diagonals and 2's in the off-diagonals to quickly specify an unstructured constraint in the Gtc argument of the vsm function.
Usage
unsm(x, reps=NULL)
Arguments
x |
integer specifying the number of traits to be fitted for a given random effect. |
reps |
integer specifying the number of times the matrix should be repeated in a list format to provide easily the constraints in complex models that use the ds(), us() or cs() structures. |
Value
- $res
a matrix or a list of matrices with the constraints to be provided in the Gtc argument of the
vsmfunction.
Author(s)
Giovanny Covarrubias-Pazaran
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Examples
unsm(3)
unsm(3,2)
unstructured covariance structure
Description
usm creates an unstructured covariance structure for specific levels of
the random effect to be used with the mmes solver.
Usage
usm(x, thetaC, theta)
Arguments
x |
vector of observations for the random effect. |
thetaC |
an optional symmetric matrix for constraints in the variance-covariance components. The symmetric matrix should have as many rows and columns as the number of levels in the factor 'x'. The values in the matrix define how the variance-covariance components should be estimated: 0: component will not be estimated 1: component will be estimated and constrained to be positive 2: component will be estimated and unconstrained 3: component will be fixed to the value provided in the theta argument |
theta |
an optional symmetric matrix for initial values of the variance-covariance components. When providing customized values, these values should be scaled with respect to the original variance. For example, to provide an initial value of 1 to a given variance component, theta would be built as: theta = matrix( 1 / var(response) ) The symmetric matrix should have as many rows and columns as the number of levels in the factor 'x'. The values in the matrix define the initial values of the variance-covariance components that will be subject to the constraints provided in thetaC. If not provided, initial values will be calculated as: diag(ncol(mm))*.05 + matrix(.1,ncol(mm),ncol(mm)) where mm is the incidence matrix for the factor 'x'. |
Value
- $res
a list with the provided vector and the variance covariance structure expected for the levels of the random effect.
Author(s)
Giovanny Covarrubias-Pazaran
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Examples
x <- as.factor(c(1:5,1:5,1:5));x
usm(x)
## how to use the theta and thetaC arguments:
# data(DT_example, package="enhancer")
# DT <- DT_example
# theta <- matrix(9:1,3,3);
# theta[lower.tri(theta)] <- t(theta)[lower.tri(theta)]
# theta # initial VCs
# thetaC <- fixm(3); thetaC # fixed VCs
# ans1 <- mmes(Yield~Env,
# random= ~ vsm( usm(Env,theta = theta,thetaC = thetaC),ism(Name) ),
# rcov= ~ units, nIters = 1,
# data=DT)
# summary(ans1)$varcomp
vpredict form of a LMM fitted with mmes
Description
vpredict method for class "mmes".
Post-analysis procedure to calculate linear combinations of variance components. Its intended use is when the variance components are either simple variances or are variances and covariances in an unstructured matrix. The functions covered are linear combinations of the variance components (for example, phenotypic variance), a ratio of two components (for example, heritabilities) and the correlation based on three components (for example, genetic correlation).
The calculations are based on the estimated variance parameters and their variance matrix as represented by the inverse of the Fisher or Average information matrix. Note that this matrix has zero values for fixed variance parameters including those near the parameter space boundary.
The transform is specified with a formula. On the left side of the formula is a name for the transformation. On the right side of the formula is a transformation specified with shortcut names like 'V1', 'V2', etc. The easiest way to identify these shortcut names is to use 'summary(object)$varcomp'. The rows of this object can referred to with shortcuts 'V1', 'V2', etc. See the example below.
Usage
vpredict(object, transform)
## S3 method for class 'mmes'
vpredict(object, transform)
Arguments
object |
a model fitted with the mmes function. |
transform |
a formula to calculate the function. |
Details
The delta method (e.g., Lynch and Walsh 1998, Appendix 1; Ver Hoef 2012) uses
a Taylor series expansion to approximate the moments of a function of
parameters. Here, a second-order Taylor series expansion is implemented to
approximate the standard error for a function of (co)variance parameters.
Partial first derivatives of the function are calculated by algorithmic
differentiation with deriv.
Though vpredict can calculate standard errors for non-linear functions
of (co)variance parameters from a fitted mmes model, it is limited
to non-linear functions constructed by mathematical operations such as the
arithmetic operators +, -, *, / and ^,
and single-variable functions such as exp and log. See
deriv for more information.
Value
dd |
the parameter and its standard error. |
Author(s)
Giovanny Covarrubias
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Lynch, M. and B. Walsh 1998. Genetics and Analysis of Quantitative Traits. Sinauer Associates, Inc., Sunderland, MA, USA.
Ver Hoef, J.M. 2012. Who invented the delta method? The American Statistician 66:124-127. DOI: 10.1080/00031305.2012.687494
See Also
Examples
####=========================================####
####=========================================####
#### EXAMPLE 1
#### simple example with univariate models
####=========================================####
####=========================================####
# data(DT_cpdata, package="enhancer")
# DT <- DT_cpdata
# GT <- GT_cpdata
# MP <- MP_cpdata
# #### create the variance-covariance matrix
# A <- A.mat(GT)
# #### look at the data and fit the model
# head(DT)
# mix1 <- mmes(Yield~1,
# random=~vsm(ism(id),Gu=A),
# data=DT)
# summary(mix1)$varcomp
# #### run the vpredict function
# vpredict(mix1, h2 ~ V1 / ( V1 + V2 ) )
variance structure specification
Description
vs DEPRECATED NOW. was the main function to build the variance-covariance structure for the random effects to be fitted in the mmer solver.
Usage
vs(..., Gu=NULL, Gti=NULL, Gtc=NULL, reorderGu=TRUE, buildGu=TRUE)
Arguments
... |
variance structure to be specified following the logic desired in the internal kronecker product. For example, if user wants to define a diagonal variance structure for the random effect 'genotypes'(g) with respect to a random effect 'environments'(e), this is:
being
One strength of sommer is the ability to specify very complex structures with as many kronecker products as desired. For example:
is equivalent to
where different covariance structures can be applied to the levels of |
Gu |
matrix with the known variance-covariance values for the levels of the u.th random effect (i.e. relationship matrix among individuals or any other known covariance matrix). If |
Gti |
matrix with dimensions t x t (t equal to number of traits) with initial values of the variance-covariance components for the random effect specified in the .... argument. If |
Gtc |
matrix with dimensions t x t (t equal to number of traits) of constraints for the variance-covariance components for the random effect specified in the ... argument according to the following rules:
In the multi-response scenario if the user doesn't specify this argument the default is to build an unstructured matrix (using the |
reorderGu |
a |
buildGu |
a |
Details
When providing initial values in the Gti argument the user has to provide scaled variance component values. The user can provide values from a previous model by accessing the sigma_scaled output from an mmer model or if an specific value is desired the user can obtain the scaled value as:
m = x/var(y)
where x is the desired initial value and y is the response variable. You can find an example in the DT_cpdata dataset.
Value
- $res
a list with all neccesary elements (incidence matrices, known var-cov structures, unknown covariance structures to be estimated and constraints) to be used in the mmer solver.
Author(s)
Giovanny Covarrubias-Pazaran
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Covarrubias-Pazaran G (2018) Software update: Moving the R package sommer to multivariate mixed models for genome-assisted prediction. doi: https://doi.org/10.1101/354639
See Also
The core function of the package: mmer
Examples
data(DT_example, package="enhancer")
DT <- DT_example
A <- A_example
## ============================ ##
## example to without structure
## ============================ ##
mix <- mmer(Yield~Env,
random= ~ vs(Name),
rcov=~ vs(units),
data=DT)
variance structure specification
Description
vsm is the main function to build the variance-covariance structure for the random effects to be fitted in the mmes solver.
Usage
vsm(..., Gu=NULL, buildGu=TRUE, meN=1, meTheta=NULL, meThetaC=NULL,
sp=FALSE, isFixed=FALSE, verbose=TRUE)
Arguments
... |
variance structure to be specified following the logic desired in the internal kronecker product. For example, if user wants to define a diagonal variance structure for the random effect 'genotypes'(g) with respect to a random effect 'environments'(e), this is:
being
One strength of sommer is the ability to specify very complex structures with as many kronecker products as desired. For example:
is equivalent to
where different covariance structures can be applied to the levels of |
Gu |
matrix with either the inverse (in case of mmes=TRUE) or raw variance-covariance (mmes=FALSE) values for the levels of the u.th random effect (e.g., the inverse of a relationship matrix among individuals or any other known inverse covariance matrix). If using an inverse remember to provide it in the right format and with attribute of being an inverse:
Where A is your original relationship matrix. If |
buildGu |
a |
meN |
number of main effects in the variance structure. Is always counted from last to first. |
meTheta |
variance covariance matrix between the main effects desired. Just to be modified if the number of main effects is greater of 1 (e.g., indirect genetic effects). |
meThetaC |
constraints for the variance covariance matrix between the main effects desired. Just to be modified if the number of main effects is greater of 1 (e.g., indirect genetic effects). |
sp |
a TRUE/FALSE statement to indicate if the VC from this structure should be multiplied by the scale parameter added in the mmes function through the addScaleParam argument in the |
isFixed |
a TRUE/FALSE statement to indicate if the vsm function is being used in the fixed part of the model. When TRUE, the function only returns the model matrix to avoid any error messages associated to returning all elements for a random effect. FALSE is the default since it is assumed to be used for a variance structure in a random effect. |
verbose |
a TRUE/FALSE statement to indicate if messages should be printed when special situations accur. For example, adding unphenotyped individuals to the incidence matrices when present in the relationship matrices. |
Details
...
Value
- $res
a list with all neccesary elements (incidence matrices, known var-cov structures, unknown covariance structures to be estimated and constraints) to be used in the mmes solver.
Author(s)
Giovanny Covarrubias-Pazaran
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Covarrubias-Pazaran G (2018) Software update: Moving the R package sommer to multivariate mixed models for genome-assisted prediction. doi: https://doi.org/10.1101/354639
See Also
The core function of the package: mmes
Examples
data(DT_example, package="enhancer")
DT <- DT_example
DT=DT[with(DT, order(Env)), ]
A <- A_example
# if using mmes=TRUE remember to provide relationship as inverse in the Gu argument
# Ai <- solve(A + diag(1e-4,ncol(A),ncol(A)))
# Ai <- as(as(as( Ai, "dMatrix"), "generalMatrix"), "CsparseMatrix")
x <- as.character(unique(DT$Name))
DT <- droplevels(DT[which(!is.na(match(DT$Name, x[1:5]))),])
## ============================ ##
## example without structure
## ============================ ##
ism(DT$Name)
mix <- mmes(Yield~Env,
random= ~ vsm(ism(Name)),
rcov=~ units,
nIters=10,
data=DT)
## ============================ ##
## example to without structure but
## using covariance among levels in the
## random effect Name
## ============================ ##
mix <- mmes(Yield~Env,
random= ~ vsm(ism(Name), Gu=A),
rcov=~ units,
nIters=10,
data=DT)
summary(mix)$varcomp
## ============================ ##
## example to use dsm() structure (DIAGONAL)
## ============================ ##
dsm(DT$Year)
mix <- mmes(Yield~Env,
random= ~ vsm(dsm(Year),ism(Name)),
rcov=~ vsm(dsm(Year),ism(units)),
nIters=10,
data=DT)
summary(mix)$varcomp
## ============================ ##
## example to use atm() structure (level-specific)
## ============================ ##
# unique(DT$Year)
# mix <- mmes(Yield~Env,
# random= ~ vsm(atm(Year,c("2011","2012")),ism(Name)),
# rcov=~ vsm(dsm(Year),ism(units)),
# data=DT)
## ============================ ##
## example to use usm() structure (UNSTRUCTURED)
## ============================ ##
usm(DT$Year)
mix <- mmes(Yield~Env,
random= ~ vsm(usm(Year),ism(Name)),
rcov=~ vsm(dsm(Year),ism(units)),
nIters = 10,
data=DT)
## ============================ ##
## example using structure in fixed effect
## (notice the isFixed argument)
## ============================ ##
mix <- mmes(Yield~ vsm(atm(Env,"CA.2011"), ism(Name), isFixed = TRUE),
rcov=~ units,
nIters=10,
data=DT)
variance structure specification
Description
vsr DEPRECATED NOW. was the main function to build the variance-covariance structure for the random effects to be fitted in the mmer solver.
Usage
vsr(..., Gu=NULL, Gti=NULL, Gtc=NULL, reorderGu=TRUE, buildGu=TRUE)
Arguments
... |
variance structure to be specified following the logic desired in the internal kronecker product. For example, if user wants to define a diagonal variance structure for the random effect 'genotypes'(g) with respect to a random effect 'environments'(e), this is:
being
One strength of sommer is the ability to specify very complex structures with as many kronecker products as desired. For example:
is equivalent to
where different covariance structures can be applied to the levels of |
Gu |
matrix with the known variance-covariance values for the levels of the u.th random effect (i.e. relationship matrix among individuals or any other known covariance matrix). If |
Gti |
matrix with dimensions t x t (t equal to number of traits) with initial values of the variance-covariance components for the random effect specified in the .... argument. If |
Gtc |
matrix with dimensions t x t (t equal to number of traits) of constraints for the variance-covariance components for the random effect specified in the ... argument according to the following rules:
In the multi-response scenario if the user doesn't specify this argument the default is to build an unstructured matrix (using the |
reorderGu |
a |
buildGu |
a |
Details
When providing initial values in the Gti argument the user has to provide scaled variance component values. The user can provide values from a previous model by accessing the sigma_scaled output from an mmer model or if an specific value is desired the user can obtain the scaled value as:
m = x/var(y)
where x is the desired initial value and y is the response variable. You can find an example in the DT_cpdata dataset.
Value
- $res
a list with all neccesary elements (incidence matrices, known var-cov structures, unknown covariance structures to be estimated and constraints) to be used in the mmer solver.
Author(s)
Giovanny Covarrubias-Pazaran
References
Covarrubias-Pazaran G (2016) Genome assisted prediction of quantitative traits using the R package sommer. PLoS ONE 11(6): doi:10.1371/journal.pone.0156744
Covarrubias-Pazaran G (2018) Software update: Moving the R package sommer to multivariate mixed models for genome-assisted prediction. doi: https://doi.org/10.1101/354639
Examples
data(DT_example, package="enhancer")
DT <- DT_example
A <- A_example
## ============================ ##
## example to without structure
## ============================ ##
mix <- mmer(Yield~Env,
random= ~ vsr(Name),
rcov=~ vsr(units),
data=DT)